Early exit crux tide-search

Philip Remes

William S Noble
--
You received this message because you are subscribed to the Google Groups "crux-users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to crux-users+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/crux-users/01aef6ba-796b-4c90-b00a-60b511edaa9b%40googlegroups.com.
Philip Remes
On Monday, November 25, 2019 at 7:24:09 AM UTC-8, Bill Noble wrote:
Hi Philip,I suspect that you have run into the threading bug that we are currently trying to track down. Can you try re-running your command with "--num-threads 1"?Bill
On Sun, Nov 24, 2019 at 10:17 PM Philip Remes <phili...@gmail.com> wrote:
Hello,--I'm new to Crux, and have been trying to analyze some raw files that were previously searched with Sequest. I'm having some trouble though, in that the program just stops in the middle of processing. In this case it was at 16%. These data are 1 Da isolation width DIA data with nominal mass accuracy MS2 analysis. I've set the Charge State in the spectrum header to 0, so that in Sequest in Proteome Discoverer the spectra would be searched as 2 and 3+. This may be relevant, because I noticed a warning message about the Spectrum 2 not having a charge state. I've attached the log and the parameters. Any ideas? I could send you a link to a raw file as well if you send me an email address, if this will help to debug the issue.WARNING: Spectrum 2 has no charge state. Calculating chargeThanksPhilip
You received this message because you are subscribed to the Google Groups "crux-users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to crux-...@googlegroups.com.
William S Noble
To unsubscribe from this group and stop receiving emails from it, send an email to crux-users+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/crux-users/68653a85-fd8a-4299-8cb7-9c700bd31282%40googlegroups.com.
Philip Remes
I’ve continued to look into the differences between analyses of a data set with Crux\Tide+Percolator compared to ProteomeDiscoverer\SequestHT+Percolator, from an experiment with 1 Da isolation width DIA data. I focused first on some of the Tide parameters like using exact-p-value as you suggested, mz fragment bin size, precursor tolerance, and making sure the modifications for the tide-index are correct (just the ol’ dynamic M+16 and static C+57). Pretty sure that these things are okay, I started looking into the differences in XCorrs and Percolator results. One observation is that for these data the Crux Percolator used 12 SVM parameters, and pretty much discriminated true and false positives just with XCorr: or that’s the conclusion I drew from the SVM weights and by plotting Q-Value versus XCorr. The PD Percolator used 33 SVM parameters, and put more emphasis on other things than XCorr which aren’t in the Crux\Percolator, like Fragment coverage and Longest Y Sequence Series. Another observation is that the XCorr values for the same peptide in the same scan are much different. The values correlate pretty well, but there’s a lot of negative Tide XCorrs, suggesting that something has is not the same between the XCorr implementations of Tide and Sequest-HT, although it could be just a translation that has no real effect. I hadn’t known that XCorr could be negative, but see that this could certainly happen with large fragment bins, if I’m interpreting Jimmy’s 2008 XCorr paper correctly. Anyways the result of all this is that if you plot the q-values or PEP for peptides in the same spectra, there’s little correspondence between the outputs from the two pipelines.
What I’m wondering is if maybe there are aspects of the PD pipeline that are more favorable to nominal mass accuracy/resolution MS2 spectra when the isolation window is not centered on the precursor peptide, and if there's a way to make Crux perform similarly. I’ve observed similar things for other search engines, for example Byonic has trouble identifying peptides in these files, but performs comparably to Sequest for DDA spectra, when the precursor is centered on the peak. MSFragger crashes when these files are used, so there appears to be something a little unexpected about them. I’ve not yet been able to finish a Crux\Comet + Percolator search, because I tend to get an error about a bad allocation, and also my computer uses so many resources during the analysis that I can’t do anything else with it, even with Comet num_threads = 1. Do you have any thoughts about these observations, or ideas about ways to optimize the Tide+Percolator results for this kind of data? I didn’t see on the Crux\Percolator page any parameters about the SVM features to be used. Perhaps there is a way to use the same ones as PD\Percolator?
Thanks
Philip
On Monday, November 25, 2019 at 11:16:20 AM UTC-8, Bill Noble wrote:
I'm glad to hear that it is running now. I would not worry about the out-of-range peaks. That is not an unusual number. The negative value for the number of candidates per scan is definitely weird. But the problem is almost certainly in the bookkeeping for producing that output, when we aggregate the counts over each spectrum in the data set. I think the difference in performance you are seeing likely relates to some other discrepancy in how the index is constructed or the search is run. You may get better (albeit markedly slower) results if you turn on the exact-p-value option or if you switch the score-function option to "both."Bill
To view this discussion on the web visit https://groups.google.com/d/msgid/crux-users/68653a85-fd8a-4299-8cb7-9c700bd31282%40googlegroups.com.
William S Noble
I’ve continued to look into the differences between analyses of a data set with Crux\Tide+Percolator compared to ProteomeDiscoverer\SequestHT+Percolator, from an experiment with 1 Da isolation width DIA data. I focused first on some of the Tide parameters like using exact-p-value as you suggested, mz fragment bin size, precursor tolerance, and making sure the modifications for the tide-index are correct (just the ol’ dynamic M+16 and static C+57). Pretty sure that these things are okay, I started looking into the differences in XCorrs and Percolator results. One observation is that for these data the Crux Percolator used 12 SVM parameters, and pretty much discriminated true and false positives just with XCorr: or that’s the conclusion I drew from the SVM weights and by plotting Q-Value versus XCorr. The PD Percolator used 33 SVM parameters, and put more emphasis on other things than XCorr which aren’t in the Crux\Percolator, like Fragment coverage and Longest Y Sequence Series.
Another observation is that the XCorr values for the same peptide in the same scan are much different. The values correlate pretty well, but there’s a lot of negative Tide XCorrs, suggesting that something has is not the same between the XCorr implementations of Tide and Sequest-HT, although it could be just a translation that has no real effect. I hadn’t known that XCorr could be negative, but see that this could certainly happen with large fragment bins, if I’m interpreting Jimmy’s 2008 XCorr paper correctly. Anyways the result of all this is that if you plot the q-values or PEP for peptides in the same spectra, there’s little correspondence between the outputs from the two pipelines.
What I’m wondering is if maybe there are aspects of the PD pipeline that are more favorable to nominal mass accuracy/resolution MS2 spectra when the isolation window is not centered on the precursor peptide, and if there's a way to make Crux perform similarly.
I’ve observed similar things for other search engines, for example Byonic has trouble identifying peptides in these files, but performs comparably to Sequest for DDA spectra, when the precursor is centered on the peak. MSFragger crashes when these files are used, so there appears to be something a little unexpected about them. I’ve not yet been able to finish a Crux\Comet + Percolator search, because I tend to get an error about a bad allocation, and also my computer uses so many resources during the analysis that I can’t do anything else with it, even with Comet num_threads = 1. Do you have any thoughts about these observations, or ideas about ways to optimize the Tide+Percolator results for this kind of data?
I didn’t see on the Crux\Percolator page any parameters about the SVM features to be used. Perhaps there is a way to use the same ones as PD\Percolator?
To unsubscribe from this group and stop receiving emails from it, send an email to crux-users+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/crux-users/94a0f92c-2b7f-4a7c-a2aa-45e22a444f77%40googlegroups.com.
Philip Remes
On Friday, December 20, 2019 at 9:00:29 AM UTC-8, Bill Noble wrote:
Hi Philip,
Thanks for your comments. My responses are interleaved below.
To view this discussion on the web visit https://groups.google.com/d/msgid/crux-users/94a0f92c-2b7f-4a7c-a2aa-45e22a444f77%40googlegroups.com.
William S Noble
Hi BillThanks for you reply.Q: Can you clarify what you mean by "nominal" here?A: ~0.7 m/z FWHM, +/- ~0.2 m/z mass accuracy. The data are not centered on any precursor, but are DIA data with 1 Da isolation width.
Q: Are you using a very large peptide database? And if so, are you running tide-search on an index created by tide-index?A: Yes the entire human fasta is used. For Tide I used your indexing pre-processing step, and then it all runs pretty fast. Jimmy gave me a hint about setting the spectrum batch size to 10000 which seemed to help for Comet as far as the program not grabbing all the computer resources, and now I have some results from Comet+Percolator now too in the attached slides.
Q: Are there specific parameters that you have in mind? I was under the impression that we support all of the parameters in the stand-alone version of Percolator at http://percolator.ms, but maybe we missed some.A: I notice now when I ran the Comet+Percolator pipeline that there are 16 Percolator parameters instead of the 12 used for the Tide+Percolator pipeline, including deltaCn which appears to be very discriminatory. Comet+Percolator produces almost as many unique peptide identifications as PD, although there are still half as many PSMs. As nearly as I can tell, the pipeline parameters I used for Comet and Tide are as similar as could be, so it appears that when Comet is the search engine, there are a 4 more features that are used. On slide 4 of the power point I uploaded, you can see the additional features that PD used (33 total), some of which seemed to be significant. I couldn't tell from the percolator.ms website or on yours how to specify the features that get used though. Is that supported? It could be interesting to try out some of these additional features with the data produced from Tide and Comet.
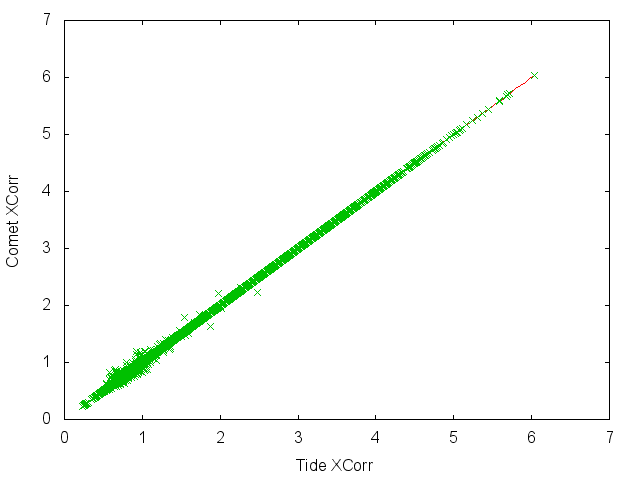
There's some additional kind of interesting observation that the Comet and Tide Xcorrs aren't exactly the same, but this may not be of real significance.
To unsubscribe from this group and stop receiving emails from it, send an email to crux-users+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/crux-users/3ae57e0c-d85f-46ec-84e7-934837d91d14%40googlegroups.com.
Philip Remes
William S Noble
No, we have not considered these other features. Not for lack of interest -- it's just something we have not had the time to do. Since many of these require access to the spectrum as well as the peptide, these features would need to be calculated by tide-search rather than make-pin. It would be relatively straightforward to add these features to the output files produced by tide-search and then modify make-pin to parse those features and include them in the Percolator input file. If this is something you are interested in trying, we could give pointers for where in the code changes should be made. But right now we are focused on some bug fixes rather than adding new functionality.Bill