When creating a gff file, should I filter out certain biotypes
70 wyświetleń
Przejdź do pierwszej nieodczytanej wiadomości

Nirad Banskota
14 wrz 2021, 15:56:2314.09.2021
do majiq_voila
Hello,
I notice that some genes have exons more than they should have. I feel that it is related to some isoforms being of the "intron retention" category. How should I filter the gtf file (and then run the gff transformation command). Should I filter out:
Intron Retention
Nonsense Mediated Decay
(Maybe processed transcript)
Or, do I leave as is?
Thanks,
Nirad
Nirad

Paul Jewell
22 wrz 2021, 12:54:4222.09.2021
do majiq_voila
Hello Nirad,
Could you give an example? Possible with the `voila view` rendering of the gene in question? Additional intron-retention should not in any case cause the creation of more exons. It is possible to have a de-novo exon, which will be easily identifiable in the rendering, however, I think the more likely cause is a use of an annotation that includes this number of exons for the genes in question. Where have you sourced the gff3 from?
Thanks.
Nirad Banskota
22 wrz 2021, 14:46:4622.09.2021
do majiq_voila
Hello Paul,
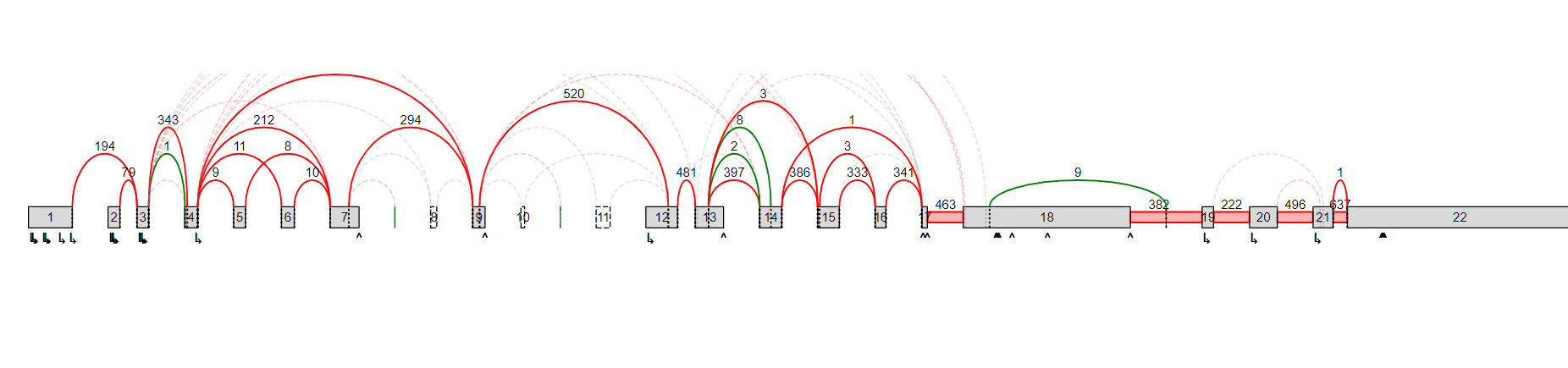
Please see attached. This gene in question (FUS) has I think 15 exons. I am seeing 19. Also, is there a way to remove denovo exons, which I am not interested in?
There is also another example that I can show you. The gene maybe has 22 or 23 exons, I am seeing 50 or so (too many denovo exons?). I would like to show that one to you as well.
The gtf file for the attached file was processed as follows:
1. MM10 ensembl gtf file was downloaded.
2. The chromosome names were converted to UCSC format from ensembl format.
3. Then I ran the gtf2gff converter.
Thank you in advance,
Nirad
{kind=link}
Nirad Banskota
22 wrz 2021, 15:02:2922.09.2021
do majiq_voila
Hello Paul,
Another example here. The gene MDM2 apparently has just 12 exons, but I am getting 22. The ensembl database was processed as follows:
1. GRCH38 version 104 (human) gtf was downloaded.
2. Again, the chromosome number was converted to UCSC format.
3. Then I ran gtf2gff tool.
Thanks,
Nirad
{kind=link}
Paul Jewell
22 wrz 2021, 15:12:3922.09.2021
do majiq_voila
Hi Nirad,
Would you be able to see Matt's answer to a similar question? https://groups.google.com/g/majiq_voila/c/miNx8SVsVHM/m/tC3dU21iAAAJ ; majiq combines exons from multiple transcripts ; this might be informative.
Nirad Banskota
22 wrz 2021, 15:32:3622.09.2021
do majiq_voila
Hello Paul,
Thank you for that link. I don't understand though, for example, how MDM2 ended up with 22 exons even after the union? Do you see the same when you use your GFF file as well?
Does it really have that many exons after taking the union of all exons?
Also, if majiq has an exon numbered as 22, how do I figure out which transcript is it coming from? Is there an ordering?
I will be using Majiq for all of my analysis, but I am trying to figure out certain things. Thank you for answering my questions. I truly appreciate it.
Thanks,
Nirad
Odpowiedz wszystkim
Odpowiedz autorowi
Przekaż
Nowe wiadomości: 0