Poor force convergence wrt basis set size
216 views
Skip to first unread message

Arthur France-Lanord
Jun 6, 2021, 6:01:31 AM6/6/21
to cp2k
Hi,
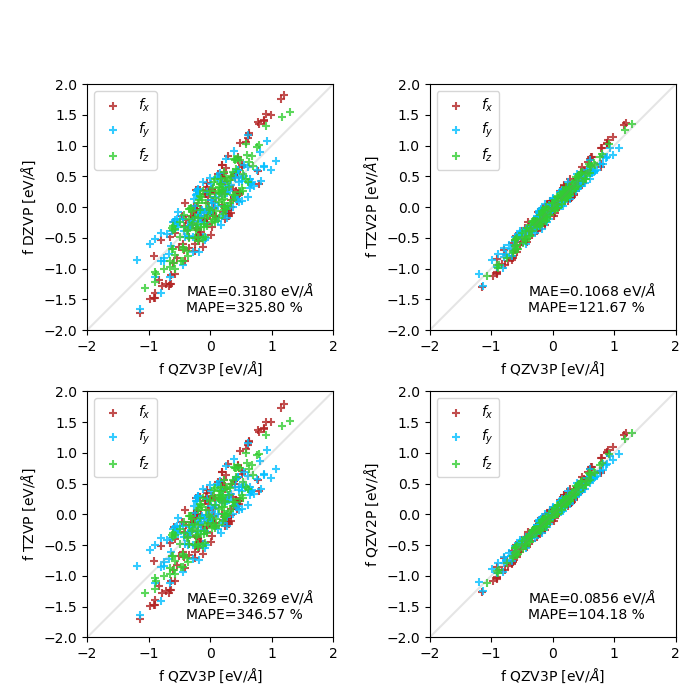
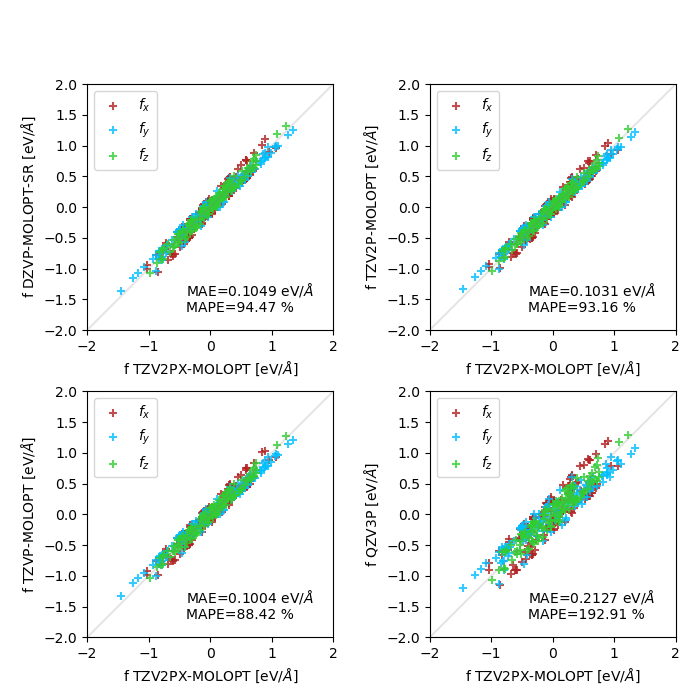
I'm currently testing atomic force convergence with respect to basis set size, for a crystalline polymorph of sulfur (an array of cyclic S8 molecules). I've tested both standard GTH basis sets (DZVP to QZV2P compared to QZV3P), and MOLOPT (DZVP-MOLOPT-SR to TZV2P-MOLOPT compared to TZV2PX-MOLOPT). I'm observing a slow or poor convergence behavior, which can be seen in these two plots (MAE : mean absolute error, MAPE : mean absolute percentage error, both only computed on the x components of the forces):
GTH basis sets:
MOLOPT basis sets (the TZV2P-MOLOPT/QZV3P panel is just shown here for comparison) :
In particular, TZVP-MOLOPT performs slightly better than TZV2P-MOLOPT..? I'm using a high energy cutoff (2500 Ry and a REL_CUTOFF of 100 Ry), and a rather tight SCF convergence criterion (1.0e-7) with the OT method. Convergence in the SCF is achieved rather fast.
Are these errors expected? I was trying to reproduce something similar from what ccan be seen in https://arxiv.org/pdf/1409.4527.pdf (figures 1 and 2), but didn't expect such high relative errors. The configuration I'm using is from a molecular dynamics run to obtain non-zero forces. I'm including a cp2k input file to reproduce the QZV3P calculation.
many thanks,
Arthur

hut...@chem.uzh.ch
Jun 16, 2021, 4:56:06 AM6/16/21
to cp...@googlegroups.com
Hi
I to be sure that you don't pick up basis set dependent numerical noise
I would suggest to use a more strict eps_default value, e.g. 1.E-14.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Arthur France-Lanord"
Sent by: cp...@googlegroups.com
Date: 06/06/2021 12:01PM
Subject: [CP2K:15521] Poor force convergence wrt basis set size
Hi,
I'm currently testing atomic force convergence with respect to basis set size, for a crystalline polymorph of sulfur (an array of cyclic S8 molecules). I've tested both standard GTH basis sets (DZVP to QZV2P compared to QZV3P), and MOLOPT (DZVP-MOLOPT-SR to TZV2P-MOLOPT compared to TZV2PX-MOLOPT). I'm observing a slow or poor convergence behavior, which can be seen in these two plots (MAE : mean absolute error, MAPE : mean absolute percentage error, both only computed on the x components of the forces):
GTH basis sets:
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/8d3ba5a1-9d0f-4dfe-8426-df09318745a9n%40googlegroups.com.
[attachment "cp2k.inp" removed by Jürg Hutter/at/UZH]
[attachment "geometry.xyz" removed by Jürg Hutter/at/UZH]
I to be sure that you don't pick up basis set dependent numerical noise
I would suggest to use a more strict eps_default value, e.g. 1.E-14.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Arthur France-Lanord"
Sent by: cp...@googlegroups.com
Date: 06/06/2021 12:01PM
Subject: [CP2K:15521] Poor force convergence wrt basis set size
Hi,
I'm currently testing atomic force convergence with respect to basis set size, for a crystalline polymorph of sulfur (an array of cyclic S8 molecules). I've tested both standard GTH basis sets (DZVP to QZV2P compared to QZV3P), and MOLOPT (DZVP-MOLOPT-SR to TZV2P-MOLOPT compared to TZV2PX-MOLOPT). I'm observing a slow or poor convergence behavior, which can be seen in these two plots (MAE : mean absolute error, MAPE : mean absolute percentage error, both only computed on the x components of the forces):
GTH basis sets:
MOLOPT basis sets (the TZV2P-MOLOPT/QZV3P panel is just shown here for comparison) :
In particular, TZVP-MOLOPT performs slightly better than TZV2P-MOLOPT..? I'm using a high energy cutoff (2500 Ry and a REL_CUTOFF of 100 Ry), and a rather tight SCF convergence criterion (1.0e-7) with the OT method. Convergence in the SCF is achieved rather fast.
Are these errors expected? I was trying to reproduce something similar from what ccan be seen in https://arxiv.org/pdf/1409.4527.pdf (figures 1 and 2), but didn't expect such high relative errors. The configuration I'm using is from a molecular dynamics run to obtain non-zero forces. I'm including a cp2k input file to reproduce the QZV3P calculation.
many thanks,
Arthur
--
Are these errors expected? I was trying to reproduce something similar from what ccan be seen in https://arxiv.org/pdf/1409.4527.pdf (figures 1 and 2), but didn't expect such high relative errors. The configuration I'm using is from a molecular dynamics run to obtain non-zero forces. I'm including a cp2k input file to reproduce the QZV3P calculation.
many thanks,
Arthur
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/8d3ba5a1-9d0f-4dfe-8426-df09318745a9n%40googlegroups.com.
[attachment "cp2k.inp" removed by Jürg Hutter/at/UZH]
[attachment "geometry.xyz" removed by Jürg Hutter/at/UZH]
Arthur France-Lanord
Jun 16, 2021, 5:47:12 AM6/16/21
to cp...@googlegroups.com
Hi,
Thank you for the suggestion. This contributed to reducing the noise on the forces, and MAE's are now ~ 30% smaller. However, I still observe almost similar performances for MOLOPT basis sets (from DZVP-MOLOPT-SR to TZV2P-MOLOPT, vs. TZV2PX-MOLOPT). In fact, the MAE is even slightly smaller for the smallest, short-range version of DZVP-MOLOPT. Could that be a symptom of over-completeness of basis sets including diffuse functions for this system?
best,
Arthur
You received this message because you are subscribed to a topic in the Google Groups "cp2k" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/cp2k/-TXuRXyRtxs/unsubscribe.
To unsubscribe from this group and all its topics, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/OF98A1FDDF.1A7050A2-ONC12586F6.003111C8-C12586F6.003111CA%40lotus.uzh.ch.
hut...@chem.uzh.ch
Jun 16, 2021, 7:35:14 AM6/16/21
to cp...@googlegroups.com
Hi
my guess (no proof available) would be that your specific system
with only S-S bonds is at the edge of the MOLOPT optimisation space.
The different variations (SR vs. orig) have been optimized for
energy and might be comparably sub-optimal for your system.
The set of S containing molecules used in the optimization is
Al2S3
CS2
H2SO4
S2
S5
SF2
HSSH
SF4
SF6
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: cp...@googlegroups.com
Date: 06/16/2021 11:47AM
Subject: Re: [CP2K:15578] Poor force convergence wrt basis set size
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAAsXd9YYVN_0R%2BeUnFh2NddACOqC68dNLptwGoX0oGhNsu1K5A%40mail.gmail.com.
my guess (no proof available) would be that your specific system
with only S-S bonds is at the edge of the MOLOPT optimisation space.
The different variations (SR vs. orig) have been optimized for
energy and might be comparably sub-optimal for your system.
The set of S containing molecules used in the optimization is
Al2S3
CS2
H2SO4
S2
S5
SF2
HSSH
SF4
SF6
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
Date: 06/16/2021 11:47AM
Subject: Re: [CP2K:15578] Poor force convergence wrt basis set size
Arthur France-Lanord
Jun 16, 2021, 11:33:08 AM6/16/21
to cp...@googlegroups.com
That sounds reasonable. It's interesting to see that DZVP-MOLOPT-SR actually performs quite well when compared to QZV3P ; on par with QZV2P.
I've been trying to use a large, generic basis set such as one from the aug-cc family, but the calculation always fails with the usual Cholesky decomposition failed message. I've tried different preconditioners, which didn't help. Do you have any tips for that? Is OT more sensitive to overcompleteness than other methods?
best,
Arthur
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/OF20A92BBB.80A39778-ONC12586F6.003F2495-C12586F6.003FA2FE%40lotus.uzh.ch.
hut...@chem.uzh.ch
Jun 16, 2021, 2:49:51 PM6/16/21
to cp...@googlegroups.com
Hi
If linear dependencies are a problem you can try
&SCF
CHOLESKY OFF
EPS_EIGVAL xxx
&END
but this will not work with OT.
There is also a new (not tested) set of basis functions that
combine the cc polariaztion and augmentation functions with
the PP valence set.
see BASIS_ccGRB_UZH in the latest TRUNK version.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: cp...@googlegroups.com
From: "Arthur France-Lanord"
Sent by: cp...@googlegroups.com
Date: 06/16/2021 05:33PM
Subject: Re: [CP2K:15581] Poor force convergence wrt basis set size
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAAsXd9a6qFdf%3D5nWUGcxLrLzHy9SneXciMDmNEa38Ec6grBcfA%40mail.gmail.com.
If linear dependencies are a problem you can try
&SCF
CHOLESKY OFF
EPS_EIGVAL xxx
&END
but this will not work with OT.
There is also a new (not tested) set of basis functions that
combine the cc polariaztion and augmentation functions with
the PP valence set.
see BASIS_ccGRB_UZH in the latest TRUNK version.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: cp...@googlegroups.com
From: "Arthur France-Lanord"
Sent by: cp...@googlegroups.com
Subject: Re: [CP2K:15581] Poor force convergence wrt basis set size
Reply all
Reply to author
Forward
0 new messages