smoothing after CRMAv2 and before CBS?
126 views
Skip to first unread message

Emilie
Dec 3, 2013, 2:53:44 PM12/3/13
to aroma-af...@googlegroups.com
Hi there,
I'm new to processing Affy SNP6 chips and so am mainly experimenting with different methods to date. I ran CRMAv2 and followed steps 1-4 from the vignette (http://aroma-project.org/vignettes/CRMAv2). For step 5, I want to do a paired analysis.
Previously I've used DNAcopy to perform CBS for other array types, and would like to follow a similar procedure, which includes smoothing prior to segmentation. Is this possible using the aroma.affymetrix package? So far I've followed the vignette for paired CNA analysis (http://aroma-project.org/vignettes/pairedTotalCopyNumberAnalysis) but haven't seen any options for smoothing.
thank you very much,
emilie

Pierre Neuvial
Dec 3, 2013, 5:26:38 PM12/3/13
to aroma. affymetrix
Hi Emilie,
It's certainly possible to do this within the Aroma framework (e.g. using the function "binnedSmoothing"). It's probably not as straightforward as running the segmentation directly, though, because this is not a typical use case.
In fact, I'm not sure why you want to perform smoothing before segmentation ? Smoothing is definitely not required before segmentation, and I would actually discourage to go this path because it will end up in a loss of resolution along the genome at the smoothing step.
Best,
Pierre
--
--
When reporting problems on aroma.affymetrix, make sure 1) to run the latest version of the package, 2) to report the output of sessionInfo() and traceback(), and 3) to post a complete code example.
You received this message because you are subscribed to the Google Groups "aroma.affymetrix" group with website http://www.aroma-project.org/.
To post to this group, send email to aroma-af...@googlegroups.com
To unsubscribe and other options, go to http://www.aroma-project.org/forum/
---
You received this message because you are subscribed to the Google Groups "aroma.affymetrix" group.
To unsubscribe from this group and stop receiving emails from it, send an email to aroma-affymetr...@googlegroups.com.
For more options, visit https://groups.google.com/groups/opt_out.
Emilie
Dec 4, 2013, 11:29:52 AM12/4/13
to aroma-af...@googlegroups.com
Hi Pierre,
Thanks for your answer. I may be wrong but I thought smoothing prior to segmentation was somewhat common. It is shown in the vignettes for DNACopy and seems to be fairly common in the literature (this approach was used in the Metabric paper for example, http://www.ncbi.nlm.nih.gov/pubmed/22522925).
I'd be interested in hearing more of your thoughts against this. Do you have an idea of how much resolution is lost by smoothing?
Emilie
Pierre Neuvial
Dec 5, 2013, 12:20:12 PM12/5/13
to aroma. affymetrix
Hi Emilie,
OK, so you are referring to the “smooth.CNA" function in the DNAcopy package, cf http://www.bioconductor.org/packages/2.13/bioc/vignettes/DNAcopy/inst/doc/DNAcopy.pdf
What this function is doing is detecting outliers (based on how far their signal value is from their neighbors) and shrink their signal values toward those of their neighbors.
This is indeed appropriate and recommended. I thought that by "smoothing" you meant performing some kind of local averaging of the original signal (e.g. using a mobile median or by binning): this I don't recommend. Sorry for the confusion.
To drop outliers, one possibility is to use the "dropSegmentationOutliers" function from the PSCBS package. See the vignettes at http://cran.fhcrc.org/web/packages/PSCBS/index.html
Another comment: since you are following the vignette for paired CNA analysis, I am guessing that you are working with tumor/normal pairs. If so, then you should use PSCBS rather than CBS for segmentation. PSCBS is an extension of CBS to segment not only total copy numbers but also allelic ratios. See the PSCBS vignette in the above URL.
Best,
Pierre
Henrik Bengtsson
Dec 5, 2013, 1:08:46 PM12/5/13
to aroma-af...@googlegroups.com
Pierre beat me to this one. Comments below...
On Thu, Dec 5, 2013 at 9:20 AM, Pierre Neuvial
<pierre....@genopole.cnrs.fr> wrote:
> Hi Emilie,
>
> OK, so you are referring to the “smooth.CNA" function in the DNAcopy
> package, cf
> http://www.bioconductor.org/packages/2.13/bioc/vignettes/DNAcopy/inst/doc/DNAcopy.pdf
>
> What this function is doing is detecting outliers (based on how far their
> signal value is from their neighbors) and shrink their signal values toward
> those of their neighbors.
>
> This is indeed appropriate and recommended. I thought that by "smoothing"
> you meant performing some kind of local averaging of the original signal
> (e.g. using a mobile median or by binning): this I don't recommend. Sorry
> for the confusion.
>
>
> To drop outliers, one possibility is to use the "dropSegmentationOutliers"
> function from the PSCBS package. See the vignettes at
> http://cran.fhcrc.org/web/packages/PSCBS/index.html
>
> Another comment: since you are following the vignette for paired CNA
> analysis, I am guessing that you are working with tumor/normal pairs. If
> so, then you should use PSCBS rather than CBS for segmentation. PSCBS is an
> extension of CBS to segment not only total copy numbers but also allelic
> ratios. See the PSCBS vignette in the above URL.
To balance this a little bit, I would say there may exist outliers in
the total copy number (TCN) signals that are so sever that they bias
the estimators/test statistic of CBS (which assumes Gaussian signals).
If one believes there are such outliers and worries that they are so
extreme that they would affect the segmentation severely, one could
either (i) drop or (ii) shrink ("smooth") them. In the vignettes of
the PSCBS package, I've last night [PSCBS (>= 0.39.8)]
corrected/clarified Section 'Dropping TCN outliers' to say the
following:
"There may be some outliers among the TCNs. In
CBS~\citep{OlshenA_etal_2004,VenkatramanOlshen_2007}, the authors
propose a method for identifying outliers and then to shrink such
values toward their neighbors ("smooth") before performing
segmentation. At the time CBS was developed it made sense to not just
to drop outliers because the resolution was low and every datapoint
was valuable. With modern technologies the resolution is much higher
and we can afford dropping such outliers, which can be done by:
> data <- dropSegmentationOutliers(data)
Dropping TCN outliers is optional."
Hope this clarifies.
Back to the original question: It is not possible to drop (or smooth)
outliers using the CbsModel() pipeline [I'll add that to the todo
list]. The easiest is to turn use the PSCBS package, where you can do
plain old single-track CBS segmentation, paired PSCBS segmentation and
also non-paired PSCBS segmentation. As Pierre says, if you have tumor
SNP data, you should look into doing parent-specific CN analysis,
which you can do either via paired or non-paired PSCBS depending on
whether you have match normals or not.
To take your allele-specific CRMAv2 and bring it into a format
recognized by the PSCBS package, see
http://aroma-project.org/vignettes/PairedPSCBS-lowlevel
/Henrik
On Thu, Dec 5, 2013 at 9:20 AM, Pierre Neuvial
<pierre....@genopole.cnrs.fr> wrote:
> Hi Emilie,
>
> OK, so you are referring to the “smooth.CNA" function in the DNAcopy
> package, cf
> http://www.bioconductor.org/packages/2.13/bioc/vignettes/DNAcopy/inst/doc/DNAcopy.pdf
>
> What this function is doing is detecting outliers (based on how far their
> signal value is from their neighbors) and shrink their signal values toward
> those of their neighbors.
>
> This is indeed appropriate and recommended. I thought that by "smoothing"
> you meant performing some kind of local averaging of the original signal
> (e.g. using a mobile median or by binning): this I don't recommend. Sorry
> for the confusion.
>
>
> To drop outliers, one possibility is to use the "dropSegmentationOutliers"
> function from the PSCBS package. See the vignettes at
> http://cran.fhcrc.org/web/packages/PSCBS/index.html
>
> Another comment: since you are following the vignette for paired CNA
> analysis, I am guessing that you are working with tumor/normal pairs. If
> so, then you should use PSCBS rather than CBS for segmentation. PSCBS is an
> extension of CBS to segment not only total copy numbers but also allelic
> ratios. See the PSCBS vignette in the above URL.
the total copy number (TCN) signals that are so sever that they bias
the estimators/test statistic of CBS (which assumes Gaussian signals).
If one believes there are such outliers and worries that they are so
extreme that they would affect the segmentation severely, one could
either (i) drop or (ii) shrink ("smooth") them. In the vignettes of
the PSCBS package, I've last night [PSCBS (>= 0.39.8)]
corrected/clarified Section 'Dropping TCN outliers' to say the
following:
"There may be some outliers among the TCNs. In
CBS~\citep{OlshenA_etal_2004,VenkatramanOlshen_2007}, the authors
propose a method for identifying outliers and then to shrink such
values toward their neighbors ("smooth") before performing
segmentation. At the time CBS was developed it made sense to not just
to drop outliers because the resolution was low and every datapoint
was valuable. With modern technologies the resolution is much higher
and we can afford dropping such outliers, which can be done by:
> data <- dropSegmentationOutliers(data)
Dropping TCN outliers is optional."
Hope this clarifies.
Back to the original question: It is not possible to drop (or smooth)
outliers using the CbsModel() pipeline [I'll add that to the todo
list]. The easiest is to turn use the PSCBS package, where you can do
plain old single-track CBS segmentation, paired PSCBS segmentation and
also non-paired PSCBS segmentation. As Pierre says, if you have tumor
SNP data, you should look into doing parent-specific CN analysis,
which you can do either via paired or non-paired PSCBS depending on
whether you have match normals or not.
To take your allele-specific CRMAv2 and bring it into a format
recognized by the PSCBS package, see
http://aroma-project.org/vignettes/PairedPSCBS-lowlevel
/Henrik
Emilie
Dec 12, 2013, 3:42:24 PM12/12/13
to aroma-af...@googlegroups.com, henrik.b...@aroma-project.org
Thank you both very much! I was indeed referring to smooth.cna, sorry about that confusion.
I've switched over to PSCBS and used the dropSegmentationOutliers- it seems to be running well. I've noticed that some of my samples have very fragmented profiles (see attached). Does this suggest poor quality data, or maybe an error in my normalization/plotting? Not all samples are like this, but it almost seems like the order of the of the probes is scrambled?
Emilie
Emilie
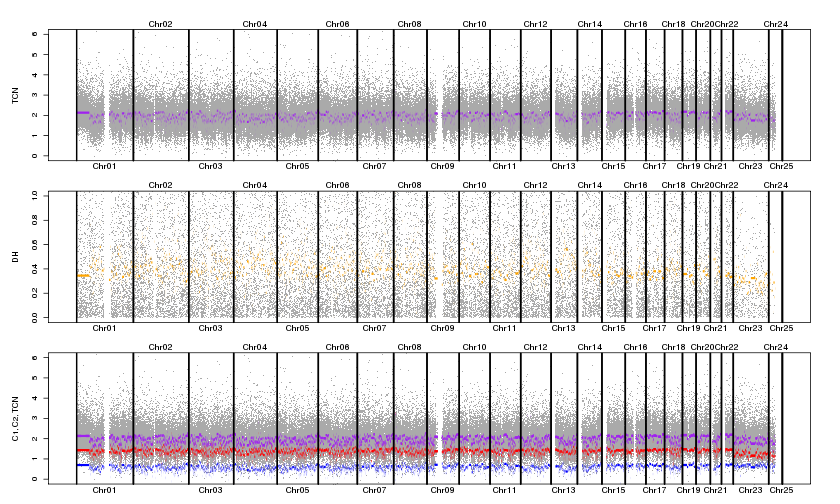{kind=link}
Henrik Bengtsson
Dec 12, 2013, 6:01:48 PM12/12/13
to aroma-affymetrix
The tumor DH panel makes me believe that either your tumor or your normal chip data is bad, or alternatively that the tumor and normal are not matched.
Check the allele B fraction of your normal. It should show three distinct bands. Do the same for the tumor. It should also show distinct bands with varying of bands depending on aberrations. If both look clean, then it's likely they're not matched. If one is very noisy, then that one is simply a bad run/sample.
Henrik
Emilie
Dec 13, 2013, 2:15:31 PM12/13/13
to aroma-af...@googlegroups.com, henrik.b...@aroma-project.org
Thanks again Henrick. I do see 3 bands, but not sure they are necessarily clean/distinct.

A similar but slightly noisier pattern is observed in the tumour sample. I also down-sampled the data 50x to be able to see the patterns (vs a black blob). Would you consider this as noise/a bad run?
Emilie
Henrik Bengtsson
Dec 13, 2013, 4:55:26 PM12/13/13
to aroma-af...@googlegroups.com
On Fri, Dec 13, 2013 at 11:15 AM, Emilie <emilie....@gmail.com> wrote:
Thanks again Henrick. I do see 3 bands, but not sure they are necessarily clean/distinct.
These normal BAFs look ok to me.
Down-sampling is alright when plotting whole-genome data.
If the tumor BAFs ('betaT') are not much noisier than the normal BAFs ('betaN'), I suspect a mismatched pair. Next, look at tumor vs normal BAFs, e.g.
plot(betaN, betaT, xlim=c(0,1), ylim=c(0,1))
What do you get? You should see most data points scattered along the diagonal line, similar to the ones in Figure 4 of the online CalMaTe vignette [ http://aroma-project.org/vignettes/CalMaTe ]. If you see data points all over the place, particularly in upper-left and the lower-right corners, your tumor normal pair is not from the same patient.
/Henrik
Emilie
Dec 16, 2013, 3:03:17 PM12/16/13
to aroma-af...@googlegroups.com, henrik.b...@aroma-project.org
Yep, I think you're right, for this sample there are many points away from the y=x line.
Two more questions that I haven't been able to find in the documentation..

Thankfully this isn't the case for all the samples though. I suppose other than finding the right match algorithmically (as this is public data), I could use a pooled reference instead of the matched normal.
Two more questions that I haven't been able to find in the documentation..
1- Is there a way to remove (or ideally prevent) very small segments? I'm finding several small (2-10 probes) segments with very high copy number (>10). I'm doubtful that they are all real.
2- I am finding very few deletions (<5 per genome, vs dozens of amplifications) in my samples. I'm calling gains/dels based on the standard deviation of the median probe log-ratio. Is this skewed distribution a common phenomena and should my threshold for deletions be more lenient than my threshold for gains?
Thanks!
Emilie
Henrik Bengtsson
Dec 16, 2013, 5:23:24 PM12/16/13
to aroma-af...@googlegroups.com
On Mon, Dec 16, 2013 at 12:03 PM, Emilie <emilie....@gmail.com> wrote:
Yep, I think you're right, for this sample there are many points away from the y=x line.
Thankfully this isn't the case for all the samples though. I suppose other than finding the right match algorithmically (as this is public data), I could use a pooled reference instead of the matched normal.
Being able to call a tumor-normal pair matched or not would be useful for QC purposes such as detecting misannotated samples as here. Shouldn't be too hard, but someone needs to do it. It's not unusual that you find misannotated samples in data sets - it happens all the time and it has been observed several times in GEO data sets (e.g. the folks behind progenetix.org/arraymap.org spend a lot of time fixing up such mistakes).
If you know there are matched normals, you should spend the time finding them, because that is so much more valuable than a global reference when dealing with parent-specific CNs. The matched normal is needed to identify the heterozygote SNPs, which are used to construct the DH track.
Two more questions that I haven't been able to find in the documentation..1- Is there a way to remove (or ideally prevent) very small segments? I'm finding several small (2-10 probes) segments with very high copy number (>10). I'm doubtful that they are all real.
This is called over segmentation. It's a known issue of several segmentation methods, including CBS (which is used by PSCBS). My view on this is that it needs to be taken care of downstream from the actual segmentation (=finding change points) - in the steps where one cluster/call segments. We are working on methods for dealing with this, but nothing is ready yet.
My best advice is to look at Section 'Pruning segmentation pro
file' in the PSCBS vignette, e.g.
fitP <- pruneByHClust(fit, h=0.25)
which merges segments with similar segment levels. The downside is that you manually have to tune 'h'. There is currently know other way to set that parameter.
2- I am finding very few deletions (<5 per genome, vs dozens of amplifications) in my samples. I'm calling gains/dels based on the standard deviation of the median probe log-ratio. Is this skewed distribution a common phenomena and should my threshold for deletions be more lenient than my threshold for gains?
First, I don't think one can make any general statements about the distribution of number of losses and gains, so I wouldn't worry too much, but I might be wrong. However, identifying where your copy neutral level is, that is, identify the ploidy of your tumor, will affect the distribution. In other words, you will get different calls if you think your tumor is diploid or tetraploid. (working on this as well)
Second, contrary to popular belief, calling gains and losses is not easy (we're working on that too as part of the problem). The problem is find find thresholds for gains and losses that are adjusted for normal contamination, other background signals, and ploidy (and even tumor heterogeneity in the worst case) - parameters that can be rather tricky to estimate reliably. I know that there methods for calling gains and losses that estimate these thresholds base on the noise level of the probes. I'm not very fond of such methods for the simple reason that two replicated runs can have identical segment means but completely different noise levels - you want to use the same calling thresholds in both cases. What to use instead? (We're working on it for the PSCBS case). For the total CN case I would use callers similar to those used by the GLAD segmentation method.
Hope this helps
Henrik
Emilie
Dec 17, 2013, 6:04:06 PM12/17/13
to aroma-af...@googlegroups.com, henrik.b...@aroma-project.org
thank you for all your help!
Reply all
Reply to author
Forward
0 new messages