Significant discontinuity in EOS curver for FeO

Kejiang Li
I am calculating the EOS for FeO with a 2*2*2 supercell. But there is always a significant discontinuity (sharp decrease) in the range of 4.3-4.4 angstrom lattice constant in the EOS curves, as shown in the figures below.
I have done the following tests:
- the convergence of the energy with different cutoffs;
- using different functionals, including PBE, HSE, and SCAN;
-including HSE with different HF fractions;
-using different magnetization and +U settings.
However, all the tests produce similar results with an apparent discontinuity (4.3-4.3 A) .
This discontinuity is not observed in the Quantum Espresso results conducted by myself and in the results of literature that generally used VASP. I believe that the CP2K program might cause this problem.
Can you give me more suggestions on how to do further tests?
Here, I also attached one sample code I used during my tests.
Thanks a lot.
Best regards,
Kejiang
The University of Science and Technology Beijing



Kejiang Li
To add one more point.
As it was said, using a reference cell might solve this problem (https://groups.google.com/g/cp2k/c/N52jLt2yAIQ/m/JAlV3KTVUtIJ). You will notice that I used &CELL_REF in my input script, which did not solve my problem.
Thanks.
Kejiang

Jürg Hutter
what is the setup for the QE calculation (cell, k-points, magnetization)?
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Kejiang Li <lyam_...@126.com>
Sent: Monday, November 20, 2023 2:26 AM
To: cp2k
Subject: [CP2K:19531] Re: Significant discontinuity in EOS curver for FeO
[cp2k_mag_u.jpg][cp2k_mag_u2.jpg]
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/f6ca6f04-88e1-47cf-aa9f-342a276d7776n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/f6ca6f04-88e1-47cf-aa9f-342a276d7776n%40googlegroups.com?utm_medium=email&utm_source=footer>.

Krack Matthias
Hi
The EOS curve for FeO (2x2x2) looks fine for me (see attached plot) using PBE and the suggested Hubbard U(eff) value of 1.9 eV for iron with CP2K.
Best
Matthias

To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit
https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759D67572D6CC24912369949FBBA%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Kejiang Li
Thanks for your reply.
The setup for QE is significantly tested in our paper: https://www.mdpi.com/1996-1944/15/23/8316/htm
As shown in the attached table.
Best regards,
Kejiang

Kejiang Li
Thanks a lot for providing the information.
Could you please provide your input file for CP2K for reference? Maybe I made a mistake somewhere.
Best regards,
Kejiang
Kejiang Li
I have read the excellent paper you provided, in which it states: "The simulations were performed with a multiplicity (2S + 1)Fe2+ = 5 for systems with a ferrous iron and (2S + 1)Fe3+ = 6 for systems with a ferric iron, respectively."
Should I set the MULTIPLICITY in cp2k to 5 for FeO system? Does MULTIPLICITY have a noticeable influence on the EOS curve?
Btw, could you please help to explain how to calucalte the MULTIPLICITY for Fe2+ or Fe3+? I got the following results from ChatGPT, which differs from yours.
Fe²⁺ (Iron(II) Ion): Loses two electrons.
- Electron configuration:
- There are 6 electrons in the 3d orbital. According to Hund's rule, the maximum multiplicity arises when the electrons are unpaired as much as possible. The 3d orbital can hold up to 10 electrons, so with 6 electrons, 4 of them can be paired, and 2 are unpaired.
- Total spin is the number of unpaired electrons divided by 2, which is .
- Multiplicity = .
- Therefore, the multiplicity for Fe²⁺ is 3.
Fe³⁺ (Iron(III) Ion): Loses three electrons.
- Electron configuration:
- There are 5 electrons in the 3d orbital. With 5 electrons, 3 can be unpaired while 2 are paired.
- Total spin is .
- Multiplicity = .
- Therefore, the multiplicity for Fe³⁺ is 4.
Best regards,
Kejiang
Kejiang Li
Here, I also attach the test results with different REL_CUTOFF and CUTOFF. This indicates that the cutoff I used is fully converged. Could anyone tell me the possible reason for the sharp decrease in energy in the EOS curves.
Thanks.
Best regards,
Kejiang

Kejiang Li
Here, I also attached the test results with different sizes of supercells. There is still a sharp decrease or discontinuity on the left side of the lowest point.
Could anyone provide any guidance?
Thanks a lot.
Best regards,

Krack Matthias
Dear Kejiang
I have added a howto page to the CP2K wiki for DFT+U calculations with an EOS calculation for cubic FeO as an example.
HTH
Matthias
From:
cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Kejiang Li <lyam_...@126.com>
Date: Wednesday, 22 November 2023 at 07:19
To: cp2k <cp...@googlegroups.com>
Subject: [CP2K:19547] Re: Significant discontinuity in EOS curver for FeO
Dear all,
Here, I also attached the test results with different sizes of supercells. There is still a sharp decrease or discontinuity on the left side of the lowest point.
Could anyone provide any guidance?
Thanks a lot.
Best regards,
Kejiang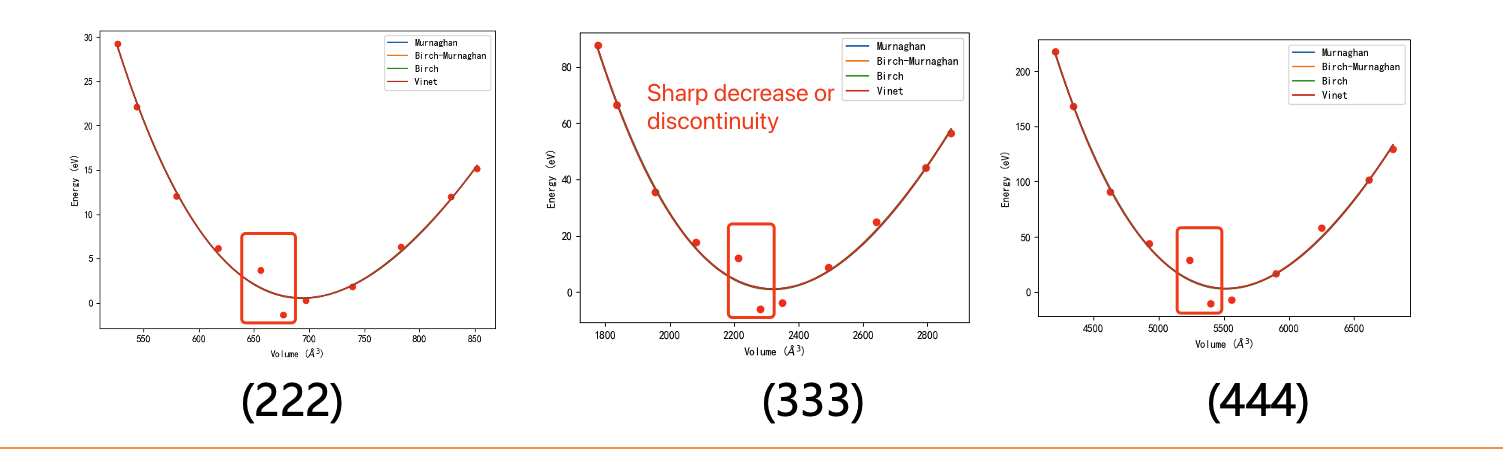
On Monday, November 20, 2023 at 9:00:11 AM UTC+8 Kejiang Li wrote:
Dear CP2K community,
I am calculating the EOS for FeO with a 2*2*2 supercell. But there is always a significant discontinuity (sharp decrease) in the range of 4.3-4.4 angstrom lattice constant in the EOS curves, as shown in the figures below.
I have done the following tests:
- the convergence of the energy with different cutoffs;
- using different functionals, including PBE, HSE, and SCAN;
-including HSE with different HF fractions;
-using different magnetization and +U settings.
However, all the tests produce similar results with an apparent discontinuity (4.3-4.3 A) .
This discontinuity is not observed in the Quantum Espresso results conducted by myself and in the results of literature that generally used VASP. I believe that the CP2K program might cause this problem.
Can you give me more suggestions on how to do further tests?
Here, I also attached one sample code I used during my tests.
Thanks a lot.
Best regards,
Kejiang
The University of Science and Technology Beijing
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to
cp2k+uns...@googlegroups.com
.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/92048ac5-9f6d-43c3-9147-d21660d5ea6an%40googlegroups.com.
Krack Matthias
Dear Kejiang
You cite a sentence describing the on-site multiplicities for each iron atom. Within an antiferromagnetic setup, e.g. for cubic FeO, the overall MULTIPLICITY of the simulation cell is 1 (the default value in CP2K with LSD).
I would not trust the answer of ChatGPT concerning the multiplicity of Fe(+2) and Fe(+3). I suggest to have a look at a text book.
Matthias
From:
cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Kejiang Li <lyam_...@126.com>
Date: Wednesday, 22 November 2023 at 05:02
To: cp2k <cp...@googlegroups.com>
Subject: Re: [CP2K:19545] Re: Significant discontinuity in EOS curver for FeO
Dear Mathias,
I have read the excellent paper you provided, in which it states: "The simulations were performed with a multiplicity (2S + 1)Fe2+ = 5 for systems with a ferrous iron and
(2S + 1)Fe3+ = 6 for systems with a ferric iron, respectively."
Should I set the MULTIPLICITY in cp2k to 5 for FeO system? Does MULTIPLICITY have a noticeable influence on the EOS curve?
Btw, could you please help to explain how to calucalte the MULTIPLICITY for Fe2+ or Fe3+? I got the following results from ChatGPT, which differs from yours.
1. Fe²⁺ (Iron(II) Ion): Loses two electrons.
· Electron configuration: [Ar]3d6
· There are 6 electrons in the 3d orbital. According to Hund's rule, the maximum multiplicity arises when the electrons are unpaired as much as possible. The 3d orbital can hold up to 10 electrons, so with 6 electrons, 4 of them can be paired, and 2 are unpaired.
· Total spin S is the number of unpaired electrons divided by 2, which is 2/2=1.
· Multiplicity = 2S+1=2×1+1=3.
· Therefore, the multiplicity for Fe²⁺ is 3.
2. Fe³⁺ (Iron(III) Ion): Loses three electrons.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/925e8f6f-1e66-48ce-ba01-659a5895c3a4n%40googlegroups.com.
Kejiang Li
Thanks a lot for providing this detailed tutorial for FeO. This will benefit a lot of CP2K users.
I have just done a fast test with your code, which produces very accurate results compared with my QE results (my spin direction is along 001, so this might cause a slight difference). See the attached figure.
I have also quickly compared your codes and my codes. I noticed the following main differences and have some quick questions:
1. You didn't set the VDW_POTENTIAL. Don't we need to consider VDW_POTENTIAL for FeO?
2. You didn't set the initial value for MAGNETIZATION. Is this also not necessary for FeO?
3. You didn't set the UKS and MULTIPLICITY. Is the default setting for UKS and MULTIPLICITY enough for FeO?
I think it might be due to one of the above reasons that caused the discontinuity in my EOS calculation. Anyway, I will do more tests to check the influence of the above factors.
Looking forward to hearing from you.
Thanks a lot for your kind help.
Best regards,
Kejiang

Krack Matthias
Dear Kejiang
- 1. You didn't set the VDW_POTENTIAL. Don't we need to consider VDW_POTENTIAL for FeO?
Not sure if that is needed for bulk FeO. Did you check the work of other authors? Did they apply such a vdW correction for bulk FeO?
- 2. You didn't set the initial value for MAGNETIZATION. Is this also not necessary for FeO?
The &BS section does the same, i.e. sets the initial on-site electronic configuration for the iron atoms, and it allows for a more detailed control.
- 3. You didn't set the UKS and MULTIPLICITY. Is the default setting for UKS and MULTIPLICITY enough for FeO?
Yes, it is for AFM FeO as I already explained.
Matthias
Error! Filename not specified.
Error! Filename not specified.Error! Filename not specified.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/92048ac5-9f6d-43c3-9147-d21660d5ea6an%40googlegroups.com.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to
cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/cbfe4a0e-b678-4ad7-809e-bd11dec37e4en%40googlegroups.com.
Kejiang Li
I really appreciate your patience in responding to my question point by point.
I researched the literature and saw no published EOS results for FeO using cp2k.
I finally found that it was the VDW_POTENTIAL that led to the significant discontinuity in my EOS curve. After eliminating the VDW_POTENTIAL potential in all my test codes, CP2K provides a continuous EOS curve that is quite normal. Please see the attached figure.
I never expected that VDW_POTENTIAL might have such an apparent lousy effect on the FeO structure. What do you think might be the possible fundamental mechanism for this issue? As I want to simulate the reaction between FeO surface and H2/H2O in the future, I think it might be necessary to include VDW_POTENTIAL.
Could you please provide your opinion regarding whether including VDW_POTENTIAL in my future calculations is necessary?
Thanks a lot.
Best,
Kejiang

Krack Matthias
Dear Kejiang
the default R_CUTOFF radius for the VDW_POTENTIAL is only 20 bohr (10.5 A) which can lead to a non-negligible noise in that energy contribution for a growing cell size, because the growth of the pair (neighbor) lists is not smooth (continuous). You can try a much larger R_CUTOFF radius, e.g. 25 A, for a smoother behavior.
I don’t think that a vdW correction is crucially needed for bulk FeO, but for FeO surfaces with H2/H2O adsorbed it makes sense most likely.
Best
Matthias
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Kejiang Li <lyam_...@126.com>
Date: Thursday, 23 November 2023 at 23:16
To: cp2k <cp...@googlegroups.com>
Subject: Re: [CP2K:19561] Re: Significant discontinuity in EOS curver for FeO
Dear Matthias,
I really appreciate your patience in responding to my question point by point.
I researched the literature and saw no published EOS results for FeO using cp2k.
I finally found that it was the VDW_POTENTIAL that led to the significant discontinuity in my EOS curve. After eliminating the VDW_POTENTIAL potential in all my test codes, CP2K provides a continuous EOS curve that is quite normal. Please see the attached
figure.
I never expected that VDW_POTENTIAL might have such an apparent lousy effect on the FeO structure. What do you think might be the possible fundamental mechanism for this issue? As I want to simulate the reaction between FeO surface and H2/H2O in the future,
I think it might be necessary to include VDW_POTENTIAL.
Could you please provide your opinion regarding whether including VDW_POTENTIAL in my future calculations is necessary?
Thanks a lot.
Best,
Kejiang

To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/9696539d-2ebb-484c-8a70-cabeedfa3e31n%40googlegroups.com.
Kejiang Li
