Dipole Moment. Help wanted.
155 views
Skip to first unread message

Alexander Kazakov
Jun 25, 2021, 9:55:24 AM6/25/21
to cp2k
Dear CP2K community,
I am simulating 64
molecules of water using DFT in NVT ensemble using PBC. I interested in
dipole moment values as a trajectory with MD step.
Here is what I have so far:
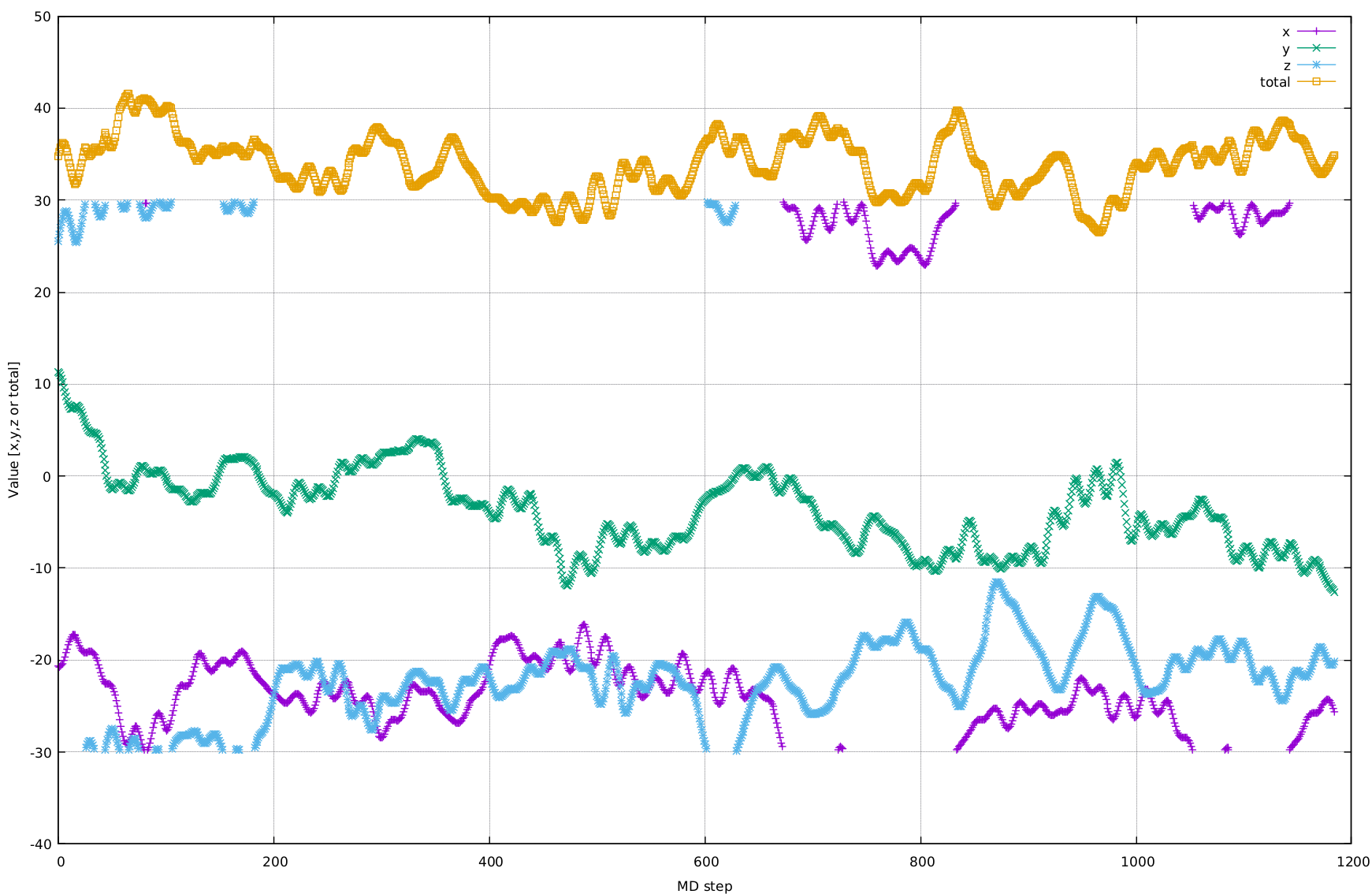
You
can see 4 curves: 3 of them are components (x,y,z) and the last one is
the length of the vector. All of them are functions of the MD step.
Let's focus on curves that correspond to x and z components. You can see that there is a kind of "jump" at certain ranges. These "jump" actually resemble a crossing kind of boundary condition. You can see how well it is fit the blue curve on the top to the bottom (the same is applied for the purple curve).
Personally, I think it is indeed a kind of boundary condition, however, I have no idea what 30 means.
Then I found these certain steps where it happens:
MD STEP
26
X= -19.18787840 Y= 5.85693184 Z= 29.55510907 Total= 35.72090144
27
X= -19.18076162 Y= 5.52351367 Z= -29.53176935 Total= 35.64458192
So the z component apparently changed the sign. So I thought that something horrible happened in the system. I plotted position atom change as a function of atom ID:
(if the picture has very poor quality, please find enclosed pdf version)
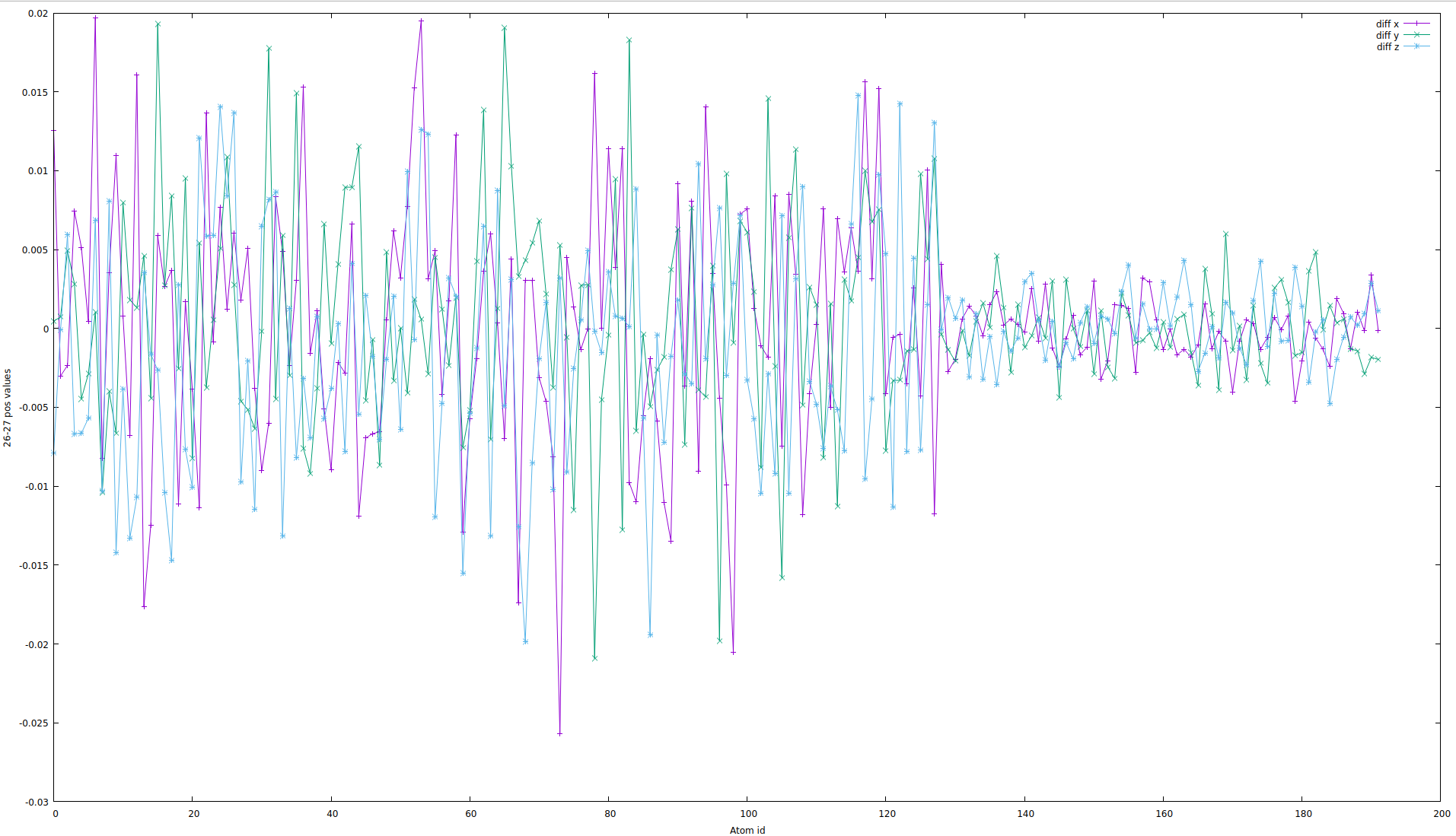
So back to questions. Could you tell me, why some components of the dipole moment "jump" changing the sigh? Is it possible to force dipole moment to stay on one side of the "boundary"? There are no doubts that hydrogen atoms should move "actively" than oxygen atoms, but I don't understand what has changed dramatically in the system. Lastly, if you can give me a hint for number 30 in the boundary (see figure), I would be thankful!
Please find enclosed FORCE_EVAL section of my script.
Additionally I am sending topology of the system.
If additionaly information is required, please let me know.
Best regards,
Alexander Kazakov
Alexander Kazakov

hut...@chem.uzh.ch
Jun 28, 2021, 4:32:08 AM6/28/21
to cp...@googlegroups.com
Hi
dipole moments in periodic systems are only defined modulo 2Pi/L
(for simple cubic systems). The value is calculated through a
complex Logarithm that can lead to results on different branches
with offsets. Have a look at the literature for more complete
discussions and more general formulas.
The choice of reference point can lead to a reduced number of
"jumps" and easier post-processing.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Alexander Kazakov"
Sent by: cp...@googlegroups.com
Date: 06/25/2021 03:58PM
Subject: [CP2K:15636] Re: Dipole Moment. Help wanted.
Yeap. Topology is here.
Best regards,
Alexander Kazakov
пятница, 25 июня 2021 г. в 15:55:24 UTC+2, Alexander Kazakov:
Dear CP2K community,
I am simulating 64 molecules of water using DFT in NVT ensemble using PBC. I interested in dipole moment values as a trajectory with MD step.
Here is what I have so far:
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/437a492d-a3df-4e73-bdb0-31dc9c1dd1f1n%40googlegroups.com.
[attachment "W64.xyz" removed by Jürg Hutter/at/UZH]
dipole moments in periodic systems are only defined modulo 2Pi/L
(for simple cubic systems). The value is calculated through a
complex Logarithm that can lead to results on different branches
with offsets. Have a look at the literature for more complete
discussions and more general formulas.
The choice of reference point can lead to a reduced number of
"jumps" and easier post-processing.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Alexander Kazakov"
Sent by: cp...@googlegroups.com
Date: 06/25/2021 03:58PM
Subject: [CP2K:15636] Re: Dipole Moment. Help wanted.
Yeap. Topology is here.
Best regards,
Alexander Kazakov
пятница, 25 июня 2021 г. в 15:55:24 UTC+2, Alexander Kazakov:
Dear CP2K community,
I am simulating 64 molecules of water using DFT in NVT ensemble using PBC. I interested in dipole moment values as a trajectory with MD step.
Here is what I have so far:
You can see 4 curves: 3 of them are components (x,y,z) and the last one is the length of the vector. All of them are functions of the MD step.
Let's focus on curves that correspond to x and z components. You can see that there is a kind of "jump" at certain ranges. These "jump" actually resemble a crossing kind of boundary condition. You can see how well it is fit the blue curve on the top to the bottom (the same is applied for the purple curve).
Personally, I think it is indeed a kind of boundary condition, however, I have no idea what 30 means.
Then I found these certain steps where it happens:
MD STEP
26
X= -19.18787840 Y= 5.85693184 Z= 29.55510907 Total= 35.72090144
27
X= -19.18076162 Y= 5.52351367 Z= -29.53176935 Total= 35.64458192
So the z component apparently changed the sign. So I thought that something horrible happened in the system. I plotted position atom change as a function of atom ID:
(if the picture has very poor quality, please find enclosed pdf version)
Let's focus on curves that correspond to x and z components. You can see that there is a kind of "jump" at certain ranges. These "jump" actually resemble a crossing kind of boundary condition. You can see how well it is fit the blue curve on the top to the bottom (the same is applied for the purple curve).
Personally, I think it is indeed a kind of boundary condition, however, I have no idea what 30 means.
Then I found these certain steps where it happens:
MD STEP
26
X= -19.18787840 Y= 5.85693184 Z= 29.55510907 Total= 35.72090144
27
X= -19.18076162 Y= 5.52351367 Z= -29.53176935 Total= 35.64458192
So the z component apparently changed the sign. So I thought that something horrible happened in the system. I plotted position atom change as a function of atom ID:
(if the picture has very poor quality, please find enclosed pdf version)
Alright, there is an interesting plot here. We see the different amplitude of change through the atom IDs. So first 2/3 of the curve correspond to hydrogen atoms displacement, whereas the last part corresponds to oxygen atoms.
So back to questions. Could you tell me, why some components of the dipole moment "jump" changing the sigh? Is it possible to force dipole moment to stay on one side of the "boundary"? There are no doubts that hydrogen atoms should move "actively" than oxygen atoms, but I don't understand what has changed dramatically in the system. Lastly, if you can give me a hint for number 30 in the boundary (see figure), I would be thankful!
As because I check CP2K 6.1, 7.1, 8.1, and 8.2 and I got the same behaviour with jumps I assume that I did something wrong or misunderstand something.
Please find enclosed FORCE_EVAL section of my script.
Additionally I am sending topology of the system.
If additionaly information is required, please let me know.
Best regards,
Alexander Kazakov
--
So back to questions. Could you tell me, why some components of the dipole moment "jump" changing the sigh? Is it possible to force dipole moment to stay on one side of the "boundary"? There are no doubts that hydrogen atoms should move "actively" than oxygen atoms, but I don't understand what has changed dramatically in the system. Lastly, if you can give me a hint for number 30 in the boundary (see figure), I would be thankful!
As because I check CP2K 6.1, 7.1, 8.1, and 8.2 and I got the same behaviour with jumps I assume that I did something wrong or misunderstand something.
Please find enclosed FORCE_EVAL section of my script.
Additionally I am sending topology of the system.
If additionaly information is required, please let me know.
Best regards,
Alexander Kazakov
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/437a492d-a3df-4e73-bdb0-31dc9c1dd1f1n%40googlegroups.com.
[attachment "W64.xyz" removed by Jürg Hutter/at/UZH]
Alexander Kazakov
Jun 29, 2021, 5:59:48 AM6/29/21
to cp2k
Dear Mr. Hutter,
thank you for your reply!
@Literature
I am reading now 2 papers:
* Nicola A. Spaldin DOI: 10.1016/j.jssc.2012.05.010 -- more about the problem of calculation dipole moment (polarization)
* Konstantin N. Kudin, Roberto Car, and Raffaele Resta DOI: 10.1063/1.2743018
Can you, please, suggest more relevant literature?
@Reference point
Are you talking about this: https://manual.cp2k.org/cp2k-8_1-branch/CP2K_INPUT/FORCE_EVAL/DFT/PRINT/MOMENTS.html#REFERENCE_POINT ?
I see there 4 points which could be a good options, however, all of them has good default values:
* REFERENCE
Default value:
ZERO
* REFERENCE_2 Default value: ZERO
* REFERENCE_POINT Default values: 0.00000000E+000 0.00000000E+000 0.00000000E+000
* REFERENCE_POINT_2 Default values: 0.00000000E+000 0.00000000E+000 0.00000000E+000
* REFERENCE_2 Default value: ZERO
* REFERENCE_POINT Default values: 0.00000000E+000 0.00000000E+000 0.00000000E+000
* REFERENCE_POINT_2 Default values: 0.00000000E+000 0.00000000E+000 0.00000000E+000
I am not sure that some others values can bring some benefits for the simple cubic system with 64 molecules (sparely distributed).
With best regards,
Alexander Kazakov
понедельник, 28 июня 2021 г. в 10:32:08 UTC+2, jgh:
hut...@chem.uzh.ch
Jul 1, 2021, 5:24:49 AM7/1/21
to cp...@googlegroups.com
Hi
there are many applications that discuss in more or less details
the technical aspects. From our group you can start with
General and efficient algorithms for obtaining maximally localized Wannier functions
G Berghold, CJ Mundy, AH Romero, J Hutter, M Parrinello
Physical Review B 61 (15), 10040 (2000)
The reference / reference point is specially helpfull if your system has a localized
source of the dipole, e.g. a polar molecule in solution. It that case you can
put the reference point close, so that nuclear and electronic contributions are
as small as possible.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Alexander Kazakov"
Sent by: cp...@googlegroups.com
Date: 06/29/2021 12:00PM
Subject: Re: [CP2K:15662] Re: Dipole Moment. Help wanted.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/3ccabbd2-55bf-4e8b-bfc1-a610a968d0a3n%40googlegroups.com.
there are many applications that discuss in more or less details
the technical aspects. From our group you can start with
General and efficient algorithms for obtaining maximally localized Wannier functions
G Berghold, CJ Mundy, AH Romero, J Hutter, M Parrinello
Physical Review B 61 (15), 10040 (2000)
The reference / reference point is specially helpfull if your system has a localized
source of the dipole, e.g. a polar molecule in solution. It that case you can
put the reference point close, so that nuclear and electronic contributions are
as small as possible.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Alexander Kazakov"
Sent by: cp...@googlegroups.com
Subject: Re: [CP2K:15662] Re: Dipole Moment. Help wanted.
Alexander Kazakov
Jul 1, 2021, 6:33:43 AM7/1/21
to cp2k
Dear Mr. Hutter,
thank you for the literature and the recommendations! I will work on it.
Best regards,
Alexander Kazakov
четверг, 1 июля 2021 г. в 11:24:49 UTC+2, jgh:
Reply all
Reply to author
Forward
0 new messages