Having difficulty with the AMBER NaCl tutorial. Getting errors and not sure how to source amber to do calculations.
75 views
Skip to first unread message

Fernando Amrein
Mar 20, 2023, 11:45:33 AM3/20/23
to westpa-users
Hello,
I'm trying to follow the basic_nacl tutorial from this article. So far I have done:
conda create -n westpa-2.0 python=3.9
conda activate westpa-2.0
conda install -c conda-forge westpa
conda install -c conda-forge westpa
I then ran init.sh, which seemed to work, but run.sh didn't. Logs are attached. Sorry if the question seems too basic but I have limited experience working with environments. I didn't seem to add any reference to Amber anywhere so I suspect that's the problem but I cannot figure out how to link to Amber or where to do so. Maybe I need to read further on environments.
Thanks in advance.

Marion
Mar 20, 2023, 1:24:39 PM3/20/23
to westpa...@googlegroups.com
Hi Fernando!
This tutorial does not use Amber, it uses openMM. So if you want to use that version just install openMM.
We also have the Amber version of this tutorial: basic_nacl_amber
If you decide to use the Amber version, you will need to specify the path to the amber.sh in the env.sh file.
Let us know if you need anything else!
Best,
Marion.
--
You received this message because you are subscribed to the Google Groups "westpa-users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to westpa-users...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/af5bde18-dba3-4940-b157-657daf66a8cbn%40googlegroups.com.
Fernando Amrein
Mar 20, 2023, 3:23:41 PM3/20/23
to westpa-users
Hi, thanks for the link. I managed to once again run init.sh but still run.sh is still not working. Doing $(which pmemd) and $(which cpptraj) gives the expected directories, and so does $AMBERHOME. Attached is once again the west.log file, as well as env.sh.
Marion
Mar 20, 2023, 3:30:39 PM3/20/23
to westpa...@googlegroups.com
Hi!
Can you also send one of the seg.log from the traj_segs file? So we can see if the problem is in the Amber dynamics or somewhere else.
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/be9ccc47-4be4-4a99-a95b-e6e5c8ef8d8an%40googlegroups.com.
Marion
Mar 20, 2023, 3:42:52 PM3/20/23
to westpa...@googlegroups.com
I agree, the Amber dynamic is working fine. Probably the error is in the calculation of the pcoord or the setting up for the next iteration. Can you send me one of your log files from the seg_logs folder?
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/0d7a92b3-bc6d-42a3-9348-e1502cd89425n%40googlegroups.com.
Marion
Mar 20, 2023, 3:55:33 PM3/20/23
to westpa...@googlegroups.com
Hi!
It's just a small error in the cpptraj script in the runseg.sh file. When using autoimage in a specific atom or residue, it is necessary to use the specific syntax. So, in that case, just change "autoimage fixed Na+" to "autoimage :1@Na+".
Thanks for pointing out that error, we are fixing it in the tutorial too!
Let me know if that works!
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/45480608-3302-4358-95fa-46530fc674d2n%40googlegroups.com.
Fernando Amrein
Mar 21, 2023, 10:40:41 AM3/21/23
to westpa...@googlegroups.com
That worked! Thanks
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/CACHGp6GgG33wsP0p9bu7QzJ9NgHUO86bwrEKu_rWVGfE-FUWSw%40mail.gmail.com.
Fernando Amrein
Mar 21, 2023, 10:56:02 AM3/21/23
to westpa...@googlegroups.com
Sorry, Im having trouble again. In the analysis section, when I run w_ipa -ao, I get the following:
Welcome to w_ipa (WESTPA Interactive Python Analysis) v. 1.0B!
Run w.introduction for a more thorough introduction, or w.help to see a list of options.
Running analysis & loading files.
Complete!
Setting iteration to iter 100.
Your current scheme, system and iteration are : TEST, /home/famrein/Downloads/westpa_tutorials-main/additional_tutorials/basic_nacl_amber, 100
Exception in thread receiver:
Traceback (most recent call last):
File "/home/famrein/anaconda3/envs/westpa-2.0/lib/python3.9/threading.py", line 980, in _bootstrap_inner
self.run()
File "/home/famrein/anaconda3/envs/westpa-2.0/lib/python3.9/threading.py", line 917, in run
self._target(*self._args, **self._kwargs)
File "/home/famrein/anaconda3/envs/westpa-2.0/lib/python3.9/site-packages/westpa/work_managers/processes.py", line 96, in results_loop
Fatal Python error: _enter_buffered_busy: could not acquire lock for <_io.BufferedWriter name='<stderr>'> at interpreter shutdown, possibly due to daemon threads
Python runtime state: finalizing (tstate=0x1999ef0)
Run w.introduction for a more thorough introduction, or w.help to see a list of options.
Running analysis & loading files.
Complete!
Setting iteration to iter 100.
Your current scheme, system and iteration are : TEST, /home/famrein/Downloads/westpa_tutorials-main/additional_tutorials/basic_nacl_amber, 100
Exception in thread receiver:
Traceback (most recent call last):
File "/home/famrein/anaconda3/envs/westpa-2.0/lib/python3.9/threading.py", line 980, in _bootstrap_inner
self.run()
File "/home/famrein/anaconda3/envs/westpa-2.0/lib/python3.9/threading.py", line 917, in run
self._target(*self._args, **self._kwargs)
File "/home/famrein/anaconda3/envs/westpa-2.0/lib/python3.9/site-packages/westpa/work_managers/processes.py", line 96, in results_loop
Fatal Python error: _enter_buffered_busy: could not acquire lock for <_io.BufferedWriter name='<stderr>'> at interpreter shutdown, possibly due to daemon threads
Python runtime state: finalizing (tstate=0x1999ef0)

Leung, Jeremy
Mar 21, 2023, 1:01:34 PM3/21/23
to westpa...@googlegroups.com
Hi Fernando,
As seen in these lines:
Complete!
Setting iteration to iter 100.
Your current scheme, system and iteration are : TEST, /home/famrein/Downloads/westpa_tutorials-main/additional_tutorials/basic_nacl_amber, 100
The w_ipa analysis has completed successfully and there are just some problems accessing stderr at the end (presumably about logging error).
These are usually computing resource-specific and it is safe to ignore that specific exception.
Best,
-- JL
--
Jeremy M. G. Leung
PhD Candidate, Chemistry
Graduate Student Researcher, Chemistry (Chong Lab)
University of Pittsburgh | 219 Parkman Avenue, Pittsburgh, PA 15260
jml...@pitt.edu | [He, Him, His]
Jeremy M. G. Leung
PhD Candidate, Chemistry
Graduate Student Researcher, Chemistry (Chong Lab)
University of Pittsburgh | 219 Parkman Avenue, Pittsburgh, PA 15260
jml...@pitt.edu | [He, Him, His]
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/CAEk19Vc-LNSY_TMikY8vHrXkYiAAbdokiDfwBFkkYLi9Qfx-MQ%40mail.gmail.com.
Fernando Amrein
Mar 21, 2023, 1:29:22 PM3/21/23
to westpa...@googlegroups.com
I understand. My question was because I couldn't see anything when opening the .h5 files with hdfview (picture attached below). I have never used this software so maybe I am doing something wrong.
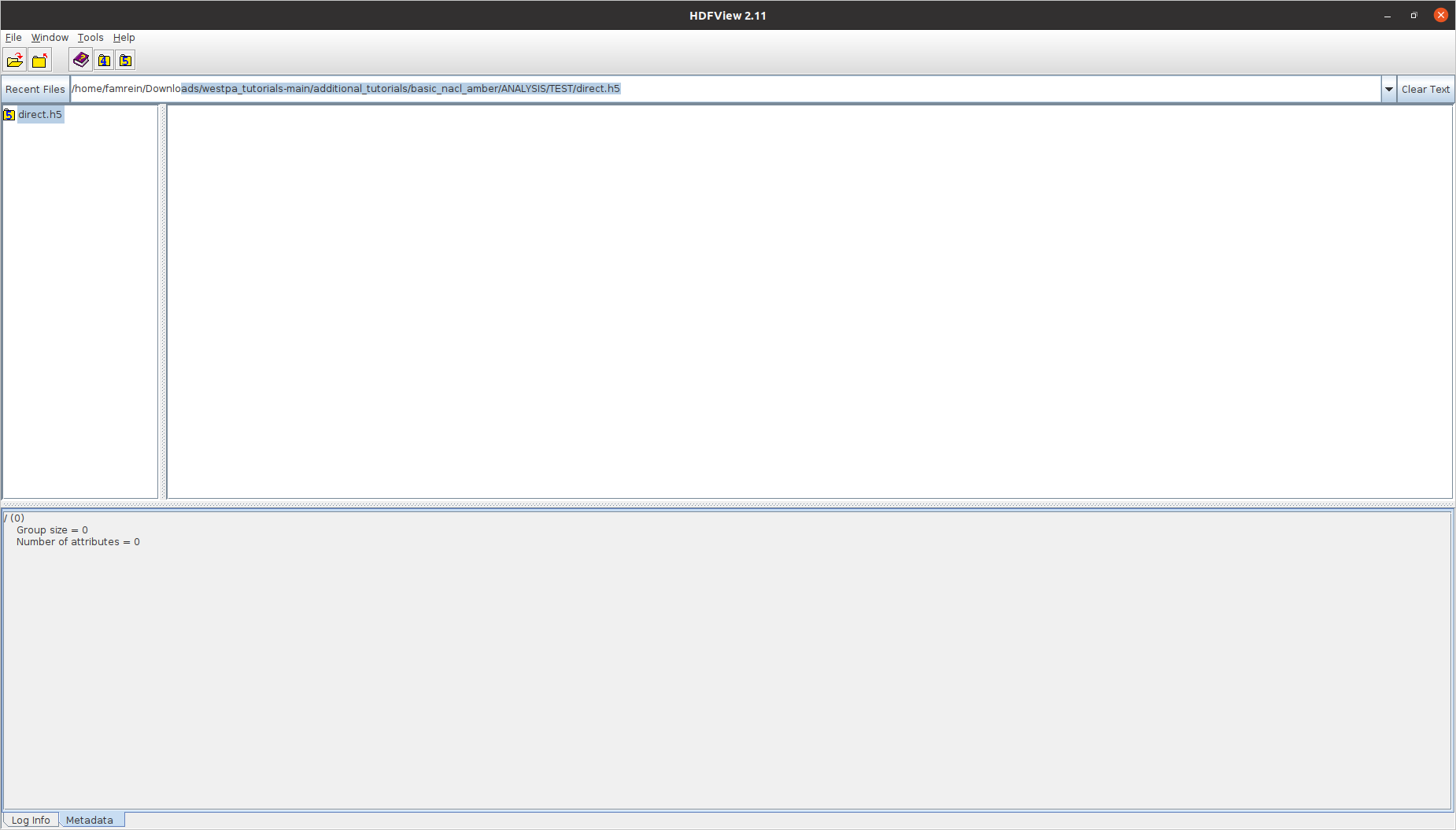
You received this message because you are subscribed to a topic in the Google Groups "westpa-users" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/westpa-users/Vzdbs2a1FcU/unsubscribe.
To unsubscribe from this group and all its topics, send an email to westpa-users...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/7774B514-8EE0-46C6-A62E-4FE0C5DE24A3%40pitt.edu.
Leung, Jeremy
Mar 21, 2023, 1:34:41 PM3/21/23
to westpa...@googlegroups.com
Hi Fernando,
You might have to download HDFView 3 in order to view the files. HDFView 2.11 is extremely old and is prone to such error.
Alternatively you can run h5py in an interactive python session (e.g. ipython) to browse the dataset.
Both options are detailed here: https://github.com/westpa/westpa/wiki/Navigating-the-west.h5-File#user-content-Install_HDFView_for_Linux_systems
-- JL
--
Jeremy M. G. Leung
PhD Candidate, Chemistry
Graduate Student Researcher, Chemistry (Chong Lab)
University of Pittsburgh | 219 Parkman Avenue, Pittsburgh, PA 15260
jml...@pitt.edu | [He, Him, His]
Jeremy M. G. Leung
PhD Candidate, Chemistry
Graduate Student Researcher, Chemistry (Chong Lab)
University of Pittsburgh | 219 Parkman Avenue, Pittsburgh, PA 15260
jml...@pitt.edu | [He, Him, His]
On 21 Mar 2023, at 1:29 PM, Fernando Amrein <fernand...@gmail.com> wrote:
I understand. My question was because I couldn't see anything when opening the .h5 files with hdfview (picture attached below). I have never used this software so maybe I am doing something wrong.
<image.png>
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/CAEk19VfBMvdyQW96G4c%3D3Mb5b8aRBkW%3DbLPwQN%2BBW09D03WS3g%40mail.gmail.com.
Fernando Amrein
Mar 21, 2023, 2:09:17 PM3/21/23
to westpa...@googlegroups.com
Under your link, I don't understand this step:
5). Allow other users to run HDFView under the folder /opt/hdfview/bin.
I am now getting an error when trying to open a file with hdfview and i think it might be related to that step. I have changed the access to read and write for "group" and "others".
To view this discussion on the web visit https://groups.google.com/d/msgid/westpa-users/D43AFE6C-2B11-47DE-8218-63B477EF0443%40pitt.edu.
Fernando Amrein
Mar 21, 2023, 2:11:39 PM3/21/23
to westpa...@googlegroups.com
For reference, other .h5 files work (for example pdist.h5).
Fernando Amrein
Mar 21, 2023, 2:20:29 PM3/21/23
to westpa...@googlegroups.com
Disregard the last two messages, it is working now. Thanks!
Reply all
Reply to author
Forward
0 new messages