Re: [TVB] Need any help for the MRI preprocessing pipeline.

WOODMAN Michael
On 4 May 2023, at 10:20, 高政源 <joygaoz...@gmail.com> wrote:
I am a newcomer to trying to use the TVB and have found the pipeline Jupyter notebook to learn the pipeline step by step. When running the TVB PIPELINE after copying them from the pubic drive to my own collab, there are any mistakes as the picture show. Waiting for your help thanks.<1683188370482.png>--
You received this message because you are subscribed to the Google Groups "TVB Users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to tvb-users+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/tvb-users/a8e9b65c-6240-40f3-9686-7210cdd08ad5n%40googlegroups.com.
<1683188370482.png>

Joey Gao
WOODMAN Michael
To view this discussion on the web visit https://groups.google.com/d/msgid/tvb-users/61bf6b53-3b3c-4e5b-86e6-f541054375adn%40googlegroups.com.
Joey Gao
1. I have pulled the containers on my computer, can I preprocess that only with mode 3 as the tvb_pipeline_v1.1 step10? From my understanding steps 1-9 are the communication steps with the cloud. Right?
I didn't find a guide to a good offline process

Michael Schirner
Joey Gao
Michael Schirner
Joey Gao
Joey Gao
Michael Schirner
Joey Gao
Thanks, I tried this way. And I believe that's the right way too.
And there's any new question.
1. I tried this pipeline with the demo dataset with the sub-CON03. And for this data, there's a /ses-postop/ path between the /sub-CON03/ and the /anat&dwi&func/. And I noticed that the path set in the converter doesn't contain the /ses-postop/ like

But my path are like

So this scipt are not suit do process for all of the data. I need to modify it to suit for my case. Right?
2. Before you share me the pipeline.sh script. I used the docker run command to generate the MRtrix3_connectome-preproc&participant two results in the mrtrix_output folder.

And I noticed using the short workaround way will save the recon-all results and It seems I must run it in this wany or I can't realize the convertion, right?.
But when I use the short workaround way. There’s a problem. Here’s my code and the error information.


Waiting for your reply. Thanks very much, again.
Joey Gao

Michael Schirner
1. Right.
2. Right, the recon-all output is needed for the subsequent conversion. I think the problem results from a change in the MRtrix3_connectome call command structure. It was changed from a one-stage call to a two-stage call structure, with "preproc" and "participant" stages, as you mentioned. To mitigate I see two strategies: First, you could replace the call
docker run ... bids/mrtrix3_connectome with either
docker run ... thevirtualbrain/tvb-pipeline-sc or with
docker run ... bids/mrtrix3_connectome:0.5.0
In the first variant you would use a snapshot of the older bids/mrtrix3_connectome container that we archived for long-term availability. Alternatively, you could specify the tag of the older container as in the second variant.
Alternatively, you could also try changing the pipeline script to work with the newer version of the bids/mrtrix3_connectome container. Here I would recommend to build up the command step-by-step by first running it with default settings as documented. The relatively complex command as needed here, may likely not be necessary anymore with the newer version.
Hope this helps!
Best,
Micha
Joey Gao
Or if it is necessary for the short workaround way to run the mrtrix processing?
Joey Gao


Michael Schirner
Joey Gao
Joey Gao
Joey Gao

I think that's a name-changing of the software update. If right, I will change the name as your pipeline to try, thanks very much.
Joey Gao

Michael Schirner
Joey Gao
Michael Schirner
Joey Gao

I'm unsure if that's a name-changing of the software update.
Michael Schirner
Joey Gao
Joey Gao
Here's a new question. Can you help me?
The test data is like this in dwi file, And it works.

But when I use my real data. There's an error making the script stopped.
The data list in dwi and the error information are as below.
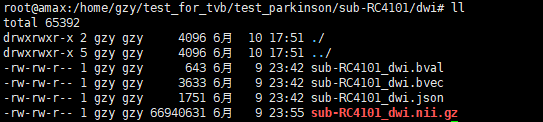

If we need the acp-AP and acp-PA data at the same time? Can I transfer my file to the right data structure. If yes, can you tell me how to do that?

Schirner, Michael
Sent: Saturday, June 10, 2023 12:48:32 PM
To: TVB Users
Subject: [ext] Re: [RESEAUX SOCIAUX] [TVB] Need any help for the MRI preprocessing pipeline.