Problems from Trimmomatic trimming
454 views
Skip to first unread message

jt...@tongji.edu.cn
Apr 23, 2017, 4:39:32 PM4/23/17
to trinityrnaseq-users
Hi,
Before using Trinity to analyze my RNA sequences, I use Trimmomatic to remove adapter sequences and bad quality parts.
My RNA sequences are paired end. I run Trimmomatic twice to remove the adapters, one with TruSeq3-PE-2.fa, one with NexteraPE-PE.fa, their adapters sequences are as follows:
TruSeq3-PE-2.fa
>PrefixPE/1
TACACTCTTTCCCTACACGACGCTCTTCCGATCT
>PrefixPE/2
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
>PE1
TACACTCTTTCCCTACACGACGCTCTTCCGATCT
>PE1_rc
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTA
>PE2
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
>PE2_rc
AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC
AGATCGGAAGAGCACACGTCAGAACTCCAGTCACAAGAGGATCTCGTATG
>TruSeq1
AGATCGGAAGAGCACACGTCAGAACTCCAGTCACACCTCAATCTCGTATG
>TruSeq2
AGATCGGAAGAGCACACGTCAGAACTCCAGTCACAGCATGATCTCGTATG
For NexteraPE-PE.fa,
>PrefixNX/1
AGATGTGTATAAGAGACAG
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCC
>PrefixNX/2
AGATGTGTATAAGAGACAG
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCC
>Trans1
TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG
>Trans1_rc
CTGTCTCTTATACACATCTGACGCTGCCGACGA
>Trans2
GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG
>Trans2_rc
CTGTCTCTTATACACATCTCCGAGCCCACGAGAC
Commands are (take NexteraPE-PE.fa as the example):
java -jar /Users/vedgcomb/Desktop/Trimmomatic-0.32/trimmomatic-0.32.jar PE 81R_S1_R1_paired.fastq 81R_S1_R2_paired.fastq
81R_R1_paired.fastq 81R_R1_unpaired.fastq 81R_R2_paired.fastq 81R_R2_unpaired.fastq ILLUMINACLIP:/Users/vedgcomb/Desktop/Trimmomatic-
0.32/adapter/NexteraPE-PE.fa:2:30:10 LEADING:20 TRAILING:20 SLIDINGWINDOW:04:24 MINLEN:50
81R_R1_paired.fastq 81R_R1_unpaired.fastq 81R_R2_paired.fastq 81R_R2_unpaired.fastq ILLUMINACLIP:/Users/vedgcomb/Desktop/Trimmomatic-
0.32/adapter/NexteraPE-PE.fa:2:30:10 LEADING:20 TRAILING:20 SLIDINGWINDOW:04:24 MINLEN:50
We can get the information reminding us it was successfully finished.
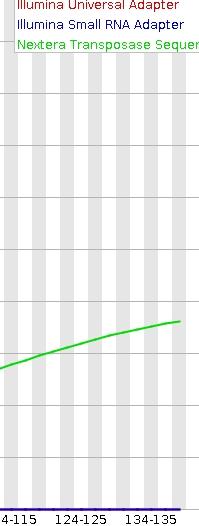
When I use the Fastqc to estimate the quality of sequences obtained after Trimmomatic-treating, it indicates that 'Adapter content ' failed (red cross) and still has much Nextera Transposase Sequences (see attachement).
Anybody can help me to analyze the reason and tell me how to do to remove the adapter sequences.
Thanks a million
Jiangtao
{kind=link}
Reply all
Reply to author
Forward
0 new messages