Unexpected zero read counts from featureCounts "-t" argument
630 views
Skip to first unread message

Chia-Yi Cheng
Feb 21, 2016, 12:01:29 AM2/21/16
to Subread
Hello All,
I was using featureCounts 1.5.0-p1 to count the reads aligned to CDS. My command was
$ featureCounts -T 8 -f -t CDS -a input.gtf -o output first.BAM second.BAM
The run completed successfully. When I reviewed the output file, I noticed many columns were zeroes and I knew many of those genes were expressed (by checking the BAM in a genome browser beforehand). For example, the output for this example locus is,
| Chr1 | 225986 | 226081 | - | 96 | 0 | 0 |
| Chr1 | 226171 | 226311 | - | 141 | 0 | 0 |
| Chr1 | 226396 | 226691 | - | 296 | 0 | 0 |
| Chr1 | 226849 | 226960 | - | 112 | 0 | 0 |
| Chr1 | 225986 | 226081 | - | 96 | 0 | 0 |
| Chr1 | 226171 | 226311 | - | 141 | 0 | 0 |
| Chr1 | 226396 | 226691 | - | 296 | 0 | 0 |
| Chr1 | 226849 | 227176 | - | 328 | 12866 | 22187 |
while the BAM is,
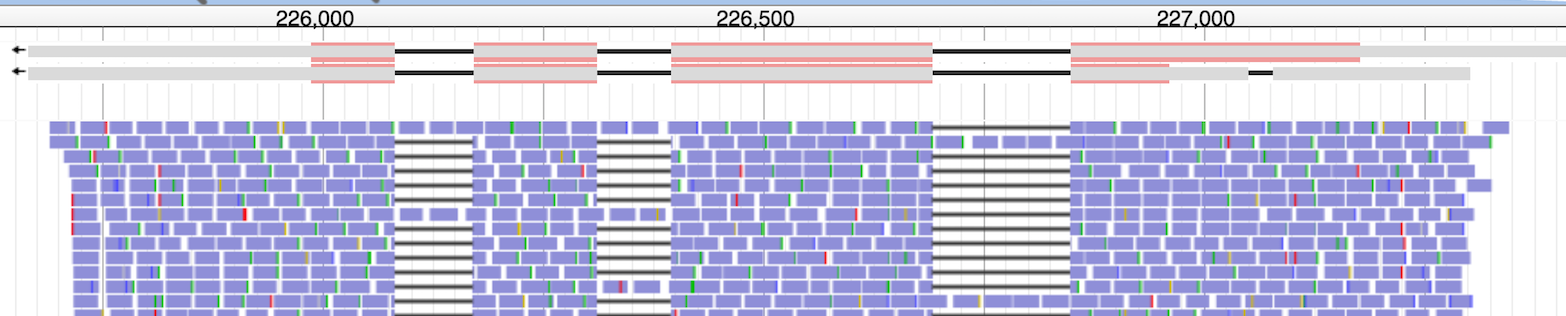
As you can see, the reads were aligned along the gene. Yet the counts were only piled in one CDS (Chr1:226849-227176).
Any comment and help is appreciated. Thank you.
Best,
Chia-Yi
ps. Here's the input GTF in this locus,
Chr1 GTF CDS 225986 226081 . - 0 transcript_id "example.2"; gene_id "example";
Chr1 GTF CDS 226171 226311 . - 0 transcript_id "example.2"; gene_id "example";
Chr1 GTF CDS 226396 226691 . - 2 transcript_id "example.2"; gene_id "example";
Chr1 GTF CDS 226849 226960 . - 0 transcript_id "example.2"; gene_id "example";
Chr1 GTF exon 226849 227049 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF 5UTR 226961 227049 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF exon 227079 227302 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF 5UTR 227079 227302 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF 3UTR 225665 225985 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF 3UTR 225665 225985 . - . transcript_id "example.1"; gene_id "example";
Chr1 GTF exon 225665 226081 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF exon 225665 226081 . - . transcript_id "example.1"; gene_id "example";
Chr1 GTF CDS 225986 226081 . - 0 transcript_id "example.1"; gene_id "example";
Chr1 GTF CDS 226171 226311 . - 0 transcript_id "example.1"; gene_id "example";
Chr1 GTF exon 226171 226311 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF exon 226171 226311 . - . transcript_id "example.1"; gene_id "example";
Chr1 GTF CDS 226396 226691 . - 2 transcript_id "example.1"; gene_id "example";
Chr1 GTF exon 226396 226691 . - . transcript_id "example.2"; gene_id "example";
Chr1 GTF exon 226396 226691 . - . transcript_id "example.1"; gene_id "example";
Chr1 GTF CDS 226849 227176 . - 0 transcript_id "example.1"; gene_id "example";
Chr1 GTF exon 226849 227543 . - . transcript_id "example.1"; gene_id "example";
Chr1 GTF 5UTR 227177 227543 . - . transcript_id "example.1"; gene_id "example";

Wei Shi
Feb 21, 2016, 5:03:39 PM2/21/16
to Subread
Hi Chia-Yi,
The reason is that your annotation includes duplicated exons (or exons overlapping with each other). FeatureCounts does not count any read that overlaps with more than one feature by default.
If you add '-O' option to your command, you will then find all your exons have counts. The '-O' option instructs featureCounts to assign a read to all its overlapping features.
Best,
Wei
The reason is that your annotation includes duplicated exons (or exons overlapping with each other). FeatureCounts does not count any read that overlaps with more than one feature by default.
If you add '-O' option to your command, you will then find all your exons have counts. The '-O' option instructs featureCounts to assign a read to all its overlapping features.
Best,
Wei
Chia-Yi Cheng
Feb 22, 2016, 11:29:26 AM2/22/16
to Subread
Hi Wei,
Thank you for the suggestion. I added '-O' and it worked as you said.
I have one follow-up question. I can see why featureCounts by default doesn't count reads for 1b as it overlaps with 1a (annotations depicted in the figure below). But I don't understand why
featureCounts did not counts reads aligned to exons 2, 3, and 4 as these exons do not overlap with 1a. Could you explain a little bit more about this?
Thank you very much!
Chia-Yi

Wei Shi
Feb 22, 2016, 5:22:39 PM2/22/16
to Subread
Hi Chia-Yi,
Take exon 2 for example, you have two copies of this exon in your annotation: 2a and 2b. For the reads mapping to exon 2, featureCounts found that they overlap with two exons and therefore it does not count them at all.
If you want to get counts for exons 2, 3 and 4, you will need to keep only one copy for each of them in your annotation.
Cheers,
Wei
Take exon 2 for example, you have two copies of this exon in your annotation: 2a and 2b. For the reads mapping to exon 2, featureCounts found that they overlap with two exons and therefore it does not count them at all.
If you want to get counts for exons 2, 3 and 4, you will need to keep only one copy for each of them in your annotation.
Cheers,
Wei
Chia-Yi Cheng
Feb 23, 2016, 3:38:55 PM2/23/16
to Subread
Hi Wei,
I understand why no reads were not counted for exon 2, 3,an 4. I am now confused by the fact that featureCounts assigned reads to exon 1a (Chr1:226849-227176 in the original post) even though exon 1a overlaps with 1b. If I understand correctly, shouldn't exon 1a get zero read counts as well?
Thank you for your time. I just wanted to make sure I completely understand '-O'.
Cheers,
Chia-Yi
Wei Shi
Feb 23, 2016, 5:08:27 PM2/23/16
to Subread
Your exon 1a overlaps with 1b, but it is not identical with 1b. The part of your exon 1a that does not overlap with 1b received counts.
Chia-Yi Cheng
Feb 25, 2016, 11:27:12 AM2/25/16
to Subread
Got it now. Thank you again for your time and answers!!
Reply all
Reply to author
Forward
0 new messages