batch effect when combining data from novaseq/hiseq runs

Oksana Vernygora
I am running stacks on combined data from 3 ddRAD runs (one
HiSeq run and two independent Novaseq runs). When samples are processed
together there’s a clear batch effect with novaseq1 samples having significant
amount of missing genotypes. I attached an IGV screenshot of the final filtered vcf file
showing those distinct blocks of samples.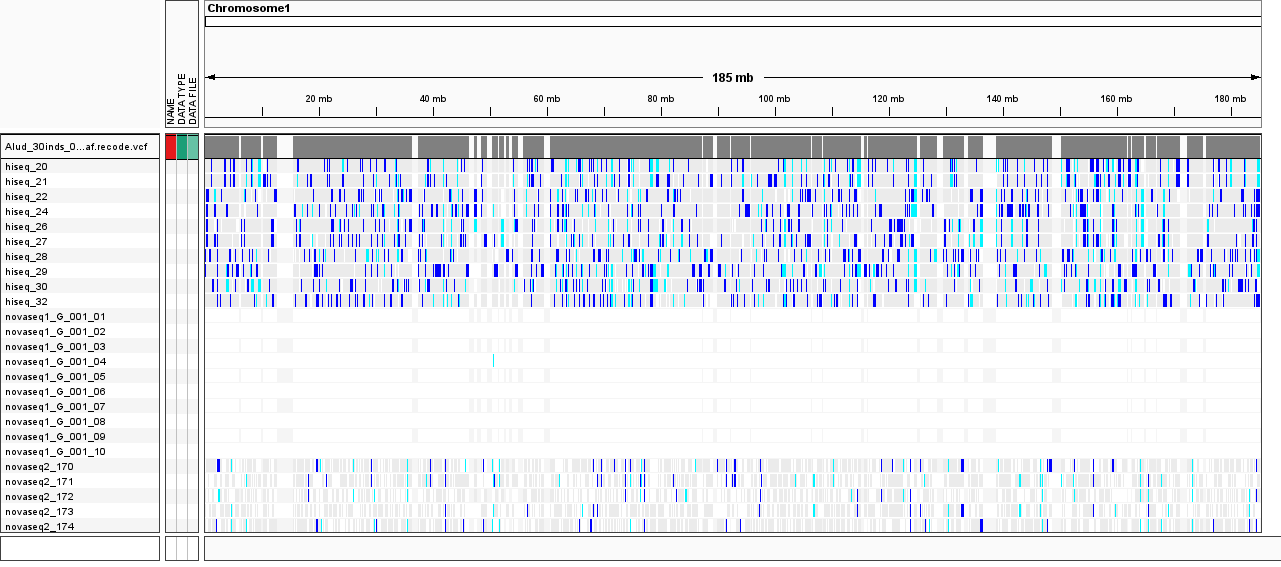
However, when I run stacks on each group of samples
separately (novaseq1, novaseq2, hiseq) with exact same parameters, this doesn't happen, e.i. novaseq1 samples genotype just fine (track_1 in the image below):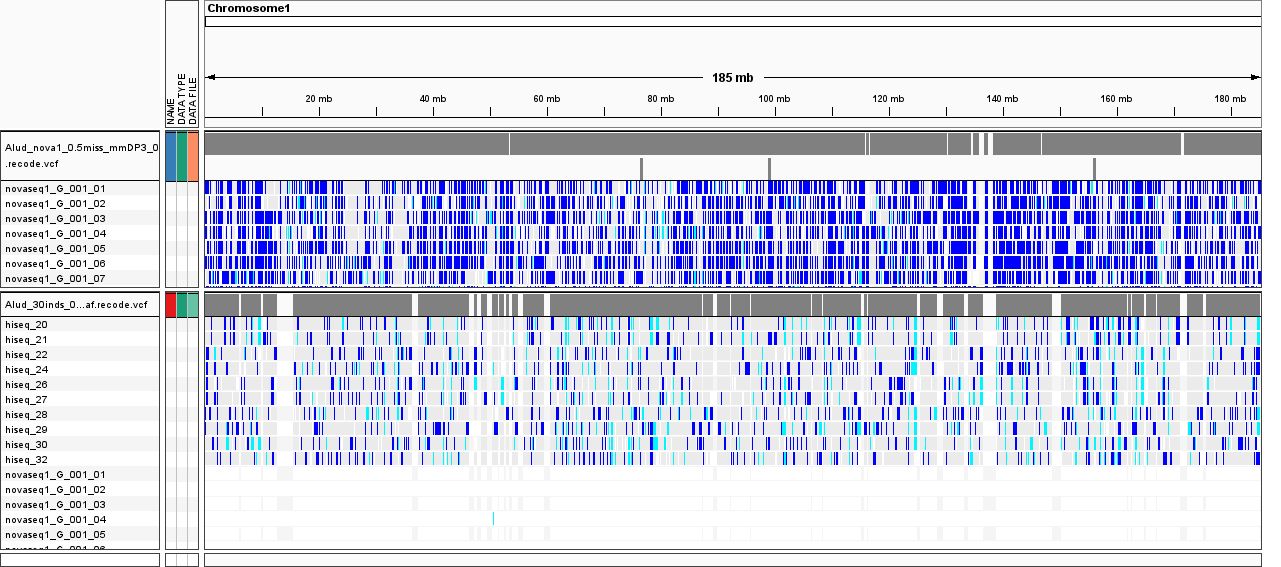
I have attached ref_map and gstacks.distributions files for these runs.
These are low coverage samples and I know that can cause issues. But I don't know what could be causing this batch effect when samples are combined.
Any insight on this issue would be greatly appreciated!
Thank you,
-Oksana
