Rescaling a selection coefficient
37 views
Skip to first unread message

Ornob Alam
Jul 28, 2021, 1:56:04 PM7/28/21
to slim-discuss
Dear Ben,
I am running a model with population sizes and generation times lowered by a factor of 10, and the mutation and recombination rates increased by the same factor.
If I wanted to simulate the trajectory of an allele with a selection coefficient of -0.2 (in a real, non-rescaled setting), rescaling makes it lower than -1. In the slim manual, there's a very nice rescaling formula for recombination rates:
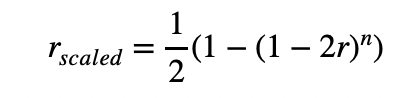
Is there something equivalent that I could do for the selection coefficient?
Lastly, I don't know if this makes a difference, but my mutation with the selection coefficient of -0.2 is recessive.
Best,
Ornob

Philipp Messer
Jul 30, 2021, 2:17:06 PM7/30/21
to Ornob Alam, slim-discuss
Hi Ornob,
Unfortunately, you are clearly in a parameter regime here where the standard rescaling approach breaks down, at least when it comes to selection. If you still want to rescale, you may have to decide which specific quantity you want to keep the same between the scaled and unscaled models.
For example, one quantity that might be useful in your case could be the fixation probability of a new mutation, which depends on its initial frequency p=1/(2N), as well as s and Ne. I think Kimura's classical formula for this probability is P(fix) = (1-exp(-4Ne*s*p))/(1-exp(-4Ne*s)). So you could try to find the s in the rescaled model that gives you the same P(fix) as in the unscaled model. Note though that this formula is for a codominant mutation, I think (but there's certainly an extension to arbitrary h somewhere).
However, there are also other things you could try to keep the same, such as the average time to loss of such a mutation, and it's not always assured that different quantities will give you the same rescaling factor for s.
Hope this helps.
Philipp
--
SLiM forward genetic simulation: http://messerlab.org/slim/
---
You received this message because you are subscribed to the Google Groups "slim-discuss" group.
To unsubscribe from this group and stop receiving emails from it, send an email to slim-discuss...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/slim-discuss/e6702863-7f70-48e8-92d2-326988ee26e9n%40googlegroups.com.
Ornob Alam
Jul 30, 2021, 4:29:03 PM7/30/21
to Philipp Messer, slim-discuss
Dear Philipp,
Thanks, that does help. I am going to try it with a couple of different quantities.
Best,
Ornob
Reply all
Reply to author
Forward
0 new messages