SLiM script repeating from a prior state of the simulation rather than asked for, losing population

Carlos Sarabia
This is my first time in this discussion group. I am excited to be able to make questions here after months of working on SLiM entirely alone! I have a problem now that I can't solve by myself.
I am simulating two populations (P1 & P2) with a 1Mb-long chromosome and 500 individuals each. Both populations accumulate slightly deleterious (S<0) and neutral mutations, but at a certain point in time, population P2 has a beneficial mutation m3 in position 500000. The simulation continues until m3 reaches fixation in P2 and then another population P3 with N=500 arises. P3 receives some migration from P2 (with mutation m3) and from P1 (without beneficial mutations). Later, the simulation continues until m3 reaches a desired frequency in P3 and outputs a VCF with all positions in P1, P2 and P3.
I want to use another software to explore its thresholds of detection of beneficial mutations in admixed populations, so I am playing with a variety of final frequencies of m3 in P3 (0-20%, 20-40%, 40-60%, 60-80%, 80-100%). Another variable that I play with is the migration proportion of population P2 into P3, from 0.5 (250 migrant individuals of P2 into P3) to 0.002 (1 migrant individual of P2 into P3). Another one is the selection coefficient of mutation m3, from highly beneficial (S=0.1) to slightly beneficial (S=0.0001).
The problem is that even though it is easy that a highly beneficial mutation (S=0.1) and a high migration proportion from pop. P2 (migp2=0.5) reaches fixation in P3, it is frequently lost in P3 if we have a low number of migrants (migp2=0.002x500 = 1 migrant) and/or if the selection coefficient is low (S=0.0001). This makes sense with population genetics theory, but I need a certain final frequency of m3 mutations in the population P3.
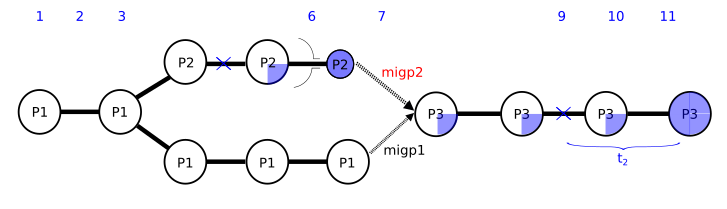
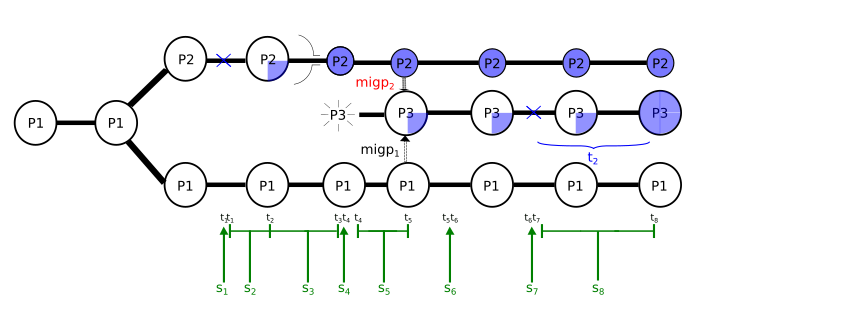
I am enclosing a scheme of the script (in a picture) and the script (below). I have subdivided the simulation in script segments defined by times so that I can play around them and be able to change them from outside using a bash command. The most important segments are s4 (between times t3 and t4), where population P3 appears, s5 (t4-t5), where P2 and P1 contribute to P3, s6 (t5-t6) where migration from P2 and P1 into P3 is closed and s8 (t7-t8) where I give some time for m3 to reach a certain frequency in pop P3. If m3 is lost in population P3, I ask the script to restart in time t7, restarting segment s8.
However the script has a problem: when mutation m3 in population P3 is lost (common if S=0.001 or if the proportion of migrants with the beneficial mutation is low), it outputs an error as if pop. P3 had not been defined (although P3 had been defined between times t3 and t4 - several generations ago!). It is as if SLiM went back generations prior to the beginning, and population P3 is erased from existence!
Changing the time to go back into t3 does not solve the problem, either. What do you think that I could do / where is the problem?
I would thank any insight. This script is driving me mad.
Best regards,
Carlos
Carlos Sarabia
Ramachandran Lab
Center of Computational Biology
Brown University
The script:
// 220822.hard sweep model in modules to calculate tfix of m3 in p3.
initialize() {
initializeSex("A");
if (exists("slimgui")) {
defineConstant("N", 500);
defineConstant("seed", 7);
defineConstant("minfreq", 0.8);
defineConstant("maxfreq", minfreq+0.2);
defineConstant("H", 0.5);
defineConstant("S0", 0.1); // S<6*10⁻5 is effectively neutral. No local hitchhiking found (Kim & Stephan, 2002)
defineConstant("S1", S0);
defineConstant("MU", 1.5e-8);
defineConstant("NMUT", 1);
defineConstant("FREQMUT", 0.0);
defineConstant("BOTTL", N*1);
defineConstant("migp2", 0.002);
defineConstant("migp1", 1-migp2);
defineConstant("TMUT", N*1);
defineConstant("times1", 5002);
defineConstant("times2", times1+5500);
defineConstant("times3", times2+1);
defineConstant("times4", times3);
defineConstant("times5", times4+1);
defineConstant("times6", times5);
defineConstant("times7", times6);
defineConstant("times8", times7+2000);
}
defineConstant("simID", getSeed());
defineConstant("direc", "/home/vant/output_slims/");
initializeMutationRate(MU); // 1.0e-8 Boyko etal 2008 // 1.5e-8 Kim,Huber & Lohmueller 2017// 1.2e-8 Durvasula & Lohmueller 2019 // 1.44e-8 Gravel etal 2013 // 1.59e-8 Ragsdale etal 2018
initializeMutationType("m1", 0.1, "g", -0.0131488, 0.186); // deleterious using Kim, Huber & Lohmueller 2017 doi: 10.1371/journal.pgen.1007741.g001
initializeMutationType("m2", 0.5, "f", 0.0); // neutral
initializeMutationType("m3", 0.5, "f", S0); // mutation from the beginning
m3.mutationStackPolicy = "f"; // m3 should not become overlapped by other mutations
m3.color = "cyan";
m3.convertToSubstitution = F; // we don't want m3 to convert to substitution.
// Coding structure (70% deleterious, 30% neutral - Eyre-Walker et al., 2007)
initializeGenomicElementType("g1", c(m1, m2), c(7,3)); // proportion 7:3 -> 70% deleterious, 30% neutral
initializeGenomicElement(g1, 0, 999999);
initializeRecombinationRate(1e-8); // Recomb. rate Messer, 2013
}
/////////////////////////////////////////////////////////////////
// First population: p1. Burn-in of 5000 generations to stabilize deleterious + neutral mutations.
1 early () { sim.addSubpop("p1", N*2); }
// Split of p2 from p1 and shrinkage of p1
5000 early() {
sim.addSubpopSplit("p2", N, p1);
p1.setSubpopulationSize(N);
}
// Whole simulation is in this event
5000 late() {
// save the state of the simulation
sim.outputFull(tempdir() + "slim_" + simID + ".txt");
// introduce the sweep mutation
// s1 - mutation m3 in p2
sim.rescheduleScriptBlock(s1, start=times1, end=times1);
// s2 - establishment of mut. P2.m3
sim.rescheduleScriptBlock(s2, start=times1, end=times2);
// s3 - bottleneck in P2
sim.rescheduleScriptBlock(s3, start=times2, end=times3);
// s4 - new pop P3
sim.rescheduleScriptBlock(s4, start=times3, end=times4);
// s5 - migration from P2 & P1 into P3
sim.rescheduleScriptBlock(s5, start=times4, end=times5);
// s6 - close migration P2+P1 -> P3
sim.rescheduleScriptBlock(s6, start=times5, end=times6);
// s7 - selection coeff of P3.m3
sim.rescheduleScriptBlock(s7, start=times6, end=times7);
// s8 - time to reach a frequency of m3 in P3
sim.rescheduleScriptBlock(s8, start=times7, end=times8);
}
// We are not interested in the bottleneck, so it is N*1
///////////////////////////////////
// Script blocks:
// s1 - mutation in p2
s1 5001 late(){
target = sample(p2.genomes, NMUT); // NMUT=number of inds with mutation
target.addNewDrawnMutation(m3, 499999);}
// s2 - establishment of mutation p2.m3
s2 5002 late() {
catn('p2.m3: ' + sim.mutationCounts(p2, sim.mutationsOfType(m3)));
if (size(sim.mutationCounts(p2, sim.mutationsOfType(m3))) == 0){
cat("LOST in p2 - RESTARTING\n");
// go back to generation in which mutation was introduced
sim.readFromPopulationFile(tempdir() + "slim_" + simID + ".txt");
sim.generation = times1;
// start a newly seeded run with a random number
setSeed(rdunif(1, 0, asInteger(2^62) - 1));
// re-introduce the sweep mutation
target = sample(p2.genomes, NMUT);
target.addNewDrawnMutation(m3, 499999);
}
if (sim.mutationCounts(p2, sim.mutationsOfType(m3)) == N*2){
cat("p2.m3 fixed\n");}
}
// s3 - bottleneck
s3 5006 early() { p2.setSubpopulationSize(asInteger(BOTTL));}
// s4 - new p3 pop
s4 5009 early() { sim.addSubpop("p3", N); catn('p3 defined'); }
// s5 - migration. p3 comes entirely from migrants -> 1-pulse migration
s5 5010 early() { p3.setMigrationRates(c(p1,p2), c(migp1,migp2));}
// s6 - close migration from p1 & p2 -> p3
s6 5011 late () { p3.setMigrationRates(c(p1,p2), c(0,0));}
// s7 - change in selection coefficient of p3.m3
s7 5015 late () { p3.genomes.mutationsOfType(m3).setSelectionCoeff(S1);}
// s8 - let time to fixation
s8 5020 late () {
FIX = asInteger(0);
gener = sim.generation; catn('generation is ' + gener);
counts = sum(p3.individuals.genomes.countOfMutationsOfType(m3));
cat("proportion of m3 alleles in p3 is " + counts/1000 + "\n");
if (counts == 0) {
cat("LOST in p3 - RESTARTING\n");
sim.readFromPopulationFile(tempdir() + "slim_" + simID + ".txt");
sim.generation = times3;
// start a newly seeded run with a random number
setSeed(rdunif(1, 0, asInteger(2^62) - 1));
}
if (((counts > N*(minfreq*2)) & (counts < N*(maxfreq*2))) | sim.generation > times8) {
cat("established beneficial mutation in p3\n");
cat("final proportion of alleles is " + counts/1000 );
genest = sim.generation;
catn('\ngeneration is ' + genest);
FIX = asInteger(sim.generation);
sim.simulationFinished();
catn("tfix: " + FIX + ".");
// we're outputing all mutations of 3 populations into a single VCF
e = p1.sampleIndividuals(N).genomes;
f = p2.sampleIndividuals(N).genomes;
g = p3.sampleIndividuals(N).genomes;
vec = c(e,f,g);
vec.outputVCF(filePath= direc + "population.p1.p2.p3.S0_" + S0 + ".migr_" + migp2 + ".S1_" + S1 + ".minfreq_" + minfreq + ".seed." + seed + ".vcf");
}
}
A normal command to run the script would be:
for i in {0001..1000}; do
slim -s $i -d seed=$i -d N=500 -d minfreq=0.8 -d maxfreq="minfreq+0.2" -d H=0.5 -d S0=0.01 -d S1="S0" -d MU=1.5e-8 -d NMUT=1 -d BOTTL="N*1" -d migp2=0.01 -d migp1="1-migp2" -d times1=5002 -d times2="times1+5500" -d times3="times2+1" -d times4="times3" -d times5="times4+1" -d times6="times5" -d times7="times6+5000" $pslim/model.slim > $fold/out.model.S0_0.01.migp2_0.01.S1_0.01.freq_0.8.$i.txt
done

Ben Haller
The advice I always give, with these dynamically rescheduled scripts, is to simplify all of that away. For debugging purposes, rewrite the script so each event is defined in the generation(s) you want it to run in, with no rescheduling. It makes it much easier to tell what's going on. In SLiMgui, it looks to me like you save out a file that contains only p1 and p2, not p3, back in tick 5000. In tick 10503 the model adds p3. In the last run I did, it got to tick 10515, noticed that the mutation had been lost, and reloaded the saved file, thus going back to the saved state with p1 and p2; this was done by your s8 script block. That script block then set the tick to 10503. The s8 script block is a late() event, so now you are in tick 10503 executing late() events. Script blocks s4 and s5 are defined as running in tick 10503, but they are early() events, so they will not be run; their scheduled time for execution has already passed. So now SLiM advanced to tick 10504, script block s5 references p3 which doesn't exist, and that's the end of the fun. I'm not sure exactly what you intended to have happen, but that's what in fact happens, and why it doesn't work. For more on exactly when script blocks get run, see section 25.11; if you don't understand why blocks s4 and s5 don't run in 10503, I think that section might clarify it. Once you get the script working the way you want it to *without* any rescheduling involved, I find that it is much easier to *then* introduce the rescheduling, by simply shifting the logic to a different base time while following the logic that you have already debugged. But working in SLiMgui also makes things much easier, since you can see, in the "object tables" window, what tick each script block has been rescheduled to, and so you can see what's happening in a way that is hard to see when running at the command line. Always debug in SLiMgui, it saves a huge amount of time. You can also see exactly what happens in each tick (events and callbacks) in SLiMgui by looking at the Scheduling tab in the debugging output window, and you can set debug points to get output at specific points in your code; these tools make it much easier to understand what's happening, particularly with dynamically rescheduled script blocks.
Good luck!
Cheers,
-B.
Benjamin C. Haller
Messer Lab
Cornell University
Carlos Sarabia
--
SLiM forward genetic simulation: http://messerlab.org/slim/
---
You received this message because you are subscribed to a topic in the Google Groups "slim-discuss" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/slim-discuss/bNkFuRPmfq0/unsubscribe.
To unsubscribe from this group and all its topics, send an email to slim-discuss...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/slim-discuss/7fd17c47-e7f8-4126-9336-75a416604a13n%40googlegroups.com.
--
Center for Computational Molecular Biology
02912 Providence, RI (USA)