Underestimate of Fis - Adding a set number of SNP
131 views
Skip to first unread message

Guillaume Lan-Fong
Nov 23, 2021, 10:31:18 AM11/23/21
to slim-discuss
Hello everyone,
I would like to ask for some help regarding an issue I have with some simulations and I don't seem to understand what is the problem. But first a little question before the "main issue".
- Adding a set number of SNPs afterwards -
First - and in order to not create a second thread for a kind of related question - I have a little question : would it be possible to create a SLiM simulation running forward, output the trees, sample some individuals and add mutations afterwards on them and their corresponding trees but with a set number S of SNP across the genomes and the trees i.e, a population of Ne individuals simulated forward during g generations of selfing, n individuals are sampled and we want the corresponding sequences and genealogies (in regard to the branches length) of these n individuals to only contain a number S of SNP.
- Underestimate Fis -
Overall context : I'm trying to simulate a quite simple population with selfing for some generations, sample a few individuals and compute the corresponding Fis.
Here are the set of parameters used for the simulations :
- Ne = 500
- µ = [1e-5, 1e-6, 1e-7, 1e-8]
- L = 100 kb
- r = 1e-6
- s = [0, 0.1, 0.2, 0.3, ... , 0.9, 1.0]
1) SLiM + recap
I first run SLiM simulations for a 2 000 generations, save the .trees corresponding files, recap and add mutations afterward with pyslim. Then, I use a python script to compute the Fis - so the pipeline is something like this : SLiM -> recap -> mutate -> Fis.
However, the observed Fis were usually a bit lower than expected, especially for high value of Fis.
However, the observed Fis were usually a bit lower than expected, especially for high value of Fis.
In order to see if something might have gone wrong with the recapitation or the "mutation" process, I then run some fully forward simulations.
2) Fully forward SLiM
This time, I first let the simulation run for 10.Ne generations without selfing before setting s to the desired value and I let the simulation run for about 2000 generations.
I then output in ms format a sample and compute the corresponding Fis - so this time, we've got something like this : SLiM -> msOutput -> Fis.
The results seems to be a bit better with this approach (which bugs me - things should be more or less the same no matter what method I'm using) but the Fis are still too low compared to the expected value.
Here's the SLiM script :
//// A HELPER FUNCTION FOR SETTING CONSTANTS THAT MIGHT BE CONFIGURED VIA COMMAND LINE.
function (void) defineCfgParam(string$ name, lifs value) {
if (!exists(name))
defineConstant(name, value);
}
// set up a simple neutral simulation
initialize() {
//initializeTreeSeq();
defineCfgParam("sim_id", "200");
defineCfgParam("Ne", 500);
defineCfgParam("mu", 1e-8);
defineCfgParam("s", 0.5);
defineCfgParam("samp", 20);
defineCfgParam("selfStart", Ne*10);
defineCfgParam("selfEnd", Ne*10+2000);
print("selfStart = " + selfStart);
print("selfEnd = " + selfEnd);
initializeMutationRate(mu);
// m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
// g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1);
// create "chromosomes" from "../parameters/rec_map.txt" file
lines = readFile("../parameters/rec_map.txt");
header = strsplit(lines[0], "\t");
if (header[0] != "chr_size"
| header[1] != "rec_rate") {
stop("Unexpected format!");
}
x = 0;
rates = NULL;
ends = NULL;
nchrs = length(lines) - 1;
for (line in lines[1:nchrs]) {
components = strsplit(line, "\t");
ends = c(ends, x + asInteger(components[0]), x + asInteger(components[0]) +1);
rates = c(rates, asFloat(components[1]), 0.5);
x = x + asInteger(components[0]) +1;
}
// remove the last base of the chr (if not, last base with 0.5 recombination rate)
rates = rates[0:(length(rates)-2)];
ends = ends[0:(length(ends)-2)];
// check the rates and ends
print(rates);
print(ends);
initializeGenomicElement(g1, 0, ends[length(ends)-1]);
initializeRecombinationRate(rates, ends);
}
// create a population of Ne individuals with selfing rate = s
1 {
sim.addSubpop("p1", Ne);
p1.setSelfingRate(0);
sim.rescheduleScriptBlock(s1, selfStart, selfStart);
sim.rescheduleScriptBlock(s2, selfEnd-1, selfEnd);
}
s1 10 late() {
print("Previous selfing rate = " + p1.selfingRate);
p1.setSelfingRate(s);
print("Generation : " + sim.generation + " - Selfing set to = " + p1.selfingRate);
}
s2 20 late() {
indGen_samp = p1.sampleIndividuals(samp).genomes;
//sim.treeSeqOutput("../results/" + asString(sim_id) + ".trees");
indGen_samp.outputMS("../results/" + asString(sim_id) + ".ms", filterMonomorphic=F);
}
function (void) defineCfgParam(string$ name, lifs value) {
if (!exists(name))
defineConstant(name, value);
}
// set up a simple neutral simulation
initialize() {
//initializeTreeSeq();
defineCfgParam("sim_id", "200");
defineCfgParam("Ne", 500);
defineCfgParam("mu", 1e-8);
defineCfgParam("s", 0.5);
defineCfgParam("samp", 20);
defineCfgParam("selfStart", Ne*10);
defineCfgParam("selfEnd", Ne*10+2000);
print("selfStart = " + selfStart);
print("selfEnd = " + selfEnd);
initializeMutationRate(mu);
// m1 mutation type: neutral
initializeMutationType("m1", 0.5, "f", 0.0);
// g1 genomic element type: uses m1 for all mutations
initializeGenomicElementType("g1", m1, 1);
// create "chromosomes" from "../parameters/rec_map.txt" file
lines = readFile("../parameters/rec_map.txt");
header = strsplit(lines[0], "\t");
if (header[0] != "chr_size"
| header[1] != "rec_rate") {
stop("Unexpected format!");
}
x = 0;
rates = NULL;
ends = NULL;
nchrs = length(lines) - 1;
for (line in lines[1:nchrs]) {
components = strsplit(line, "\t");
ends = c(ends, x + asInteger(components[0]), x + asInteger(components[0]) +1);
rates = c(rates, asFloat(components[1]), 0.5);
x = x + asInteger(components[0]) +1;
}
// remove the last base of the chr (if not, last base with 0.5 recombination rate)
rates = rates[0:(length(rates)-2)];
ends = ends[0:(length(ends)-2)];
// check the rates and ends
print(rates);
print(ends);
initializeGenomicElement(g1, 0, ends[length(ends)-1]);
initializeRecombinationRate(rates, ends);
}
// create a population of Ne individuals with selfing rate = s
1 {
sim.addSubpop("p1", Ne);
p1.setSelfingRate(0);
sim.rescheduleScriptBlock(s1, selfStart, selfStart);
sim.rescheduleScriptBlock(s2, selfEnd-1, selfEnd);
}
s1 10 late() {
print("Previous selfing rate = " + p1.selfingRate);
p1.setSelfingRate(s);
print("Generation : " + sim.generation + " - Selfing set to = " + p1.selfingRate);
}
s2 20 late() {
indGen_samp = p1.sampleIndividuals(samp).genomes;
//sim.treeSeqOutput("../results/" + asString(sim_id) + ".trees");
indGen_samp.outputMS("../results/" + asString(sim_id) + ".ms", filterMonomorphic=F);
}
Here's a plot of the results :
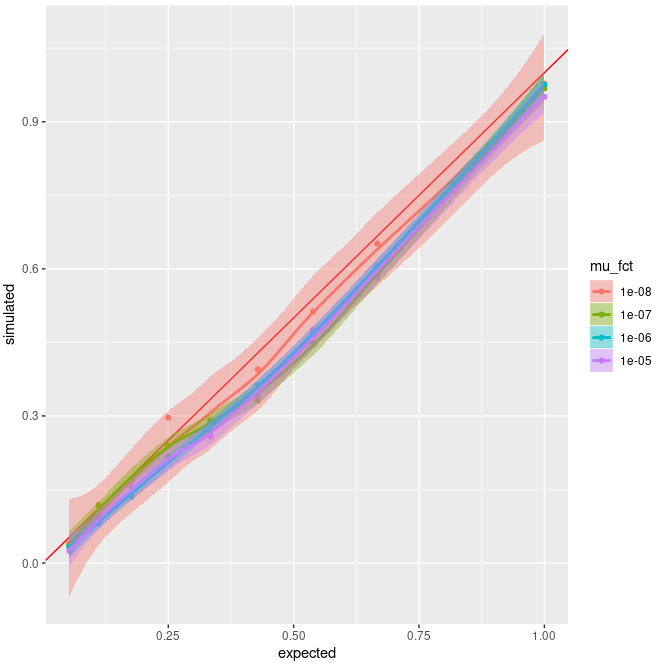
x = expected Fis, y = simulated Fis. Different colors show different value of µ. Dots are the average value of Fis. The red line is the "perfect" simulated = expected case.
So it seems that a lower mutation rate would be better, but my issue is more that I don't understand why I have an underestimation of Fis ? It might be an issue with my SLiM script itself that cause this, but having the same results with 2 different approaches seems a bit strange to me.
Once again, thanks in advance for your time and your answers,
G. L-F

Miguel de Navascués
Nov 23, 2021, 12:46:19 PM11/23/21
to slim-discuss
Hi Guillaume,
You did not specify how do you calculate expected Fis. I guess you are
using Fis=s/(2-s). That is the expected value for an equilibrium model.
None of the models you present seem to to be in equilibrium (with
recapitation or forward period without selfing). Try simulating 20*N
generation in forward with the desired selfing rate from the beginning,
I think this should get you observed Fis values around to the expectation.
Best,
Miguel
On 23/11/2021 16:31, Guillaume Lan-Fong wrote:
> Hello everyone,
>
> I would like to ask for some help regarding an issue I have with some
> simulations and I don't seem to understand what is the problem. But
> first a little question before the "main issue".
>
> *- Adding a set number of SNPs afterwards -*
>
> *- Underestimate Fis - *
>
> *Overall context* : I'm trying to simulate a quite simple population
>
> * Ne = 500
> * µ = [1e-5, 1e-6, 1e-7, 1e-8]
> * L = 100 kb
> * r = 1e-6
> * s = [0, 0.1, 0.2, 0.3, ... , 0.9, 1.0]
>
>
> *1) SLiM + recap*
>
> However, *the observed Fis were usually a bit lower than expected,
> especially for high value of Fis*.
> *
> *2) Fully forward SLiM*
>
> Here's the *SLiM script* :
> *Here's a plot of the results* :
>
> Fis_too_low.png
>
> /x = expected Fis, y = simulated Fis. Different colors show different
> SLiM forward genetic simulation: http://messerlab.org/slim/
> <http://messerlab.org/slim/>
> ---
> You received this message because you are subscribed to the Google
> Groups "slim-discuss" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to slim-discuss...@googlegroups.com
> <mailto:slim-discuss...@googlegroups.com>.
> To view this discussion on the web visit
> https://groups.google.com/d/msgid/slim-discuss/c646452e-ef1d-4825-97ce-69c1e0b054e0n%40googlegroups.com
> <https://groups.google.com/d/msgid/slim-discuss/c646452e-ef1d-4825-97ce-69c1e0b054e0n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
Miguel de Navascués
UMR CBGP, INRAE
Centre de Biologie pour la Gestion des Populations
755 avenue du campus Agropolis
CS30016
34988 Montferrier-sur-Lez cedex (France)
phone: +33499623370
fax: +33499623345
e-mail: miguel.navascues AT inrae.fr
You did not specify how do you calculate expected Fis. I guess you are
using Fis=s/(2-s). That is the expected value for an equilibrium model.
None of the models you present seem to to be in equilibrium (with
recapitation or forward period without selfing). Try simulating 20*N
generation in forward with the desired selfing rate from the beginning,
I think this should get you observed Fis values around to the expectation.
Best,
Miguel
On 23/11/2021 16:31, Guillaume Lan-Fong wrote:
> Hello everyone,
>
> I would like to ask for some help regarding an issue I have with some
> simulations and I don't seem to understand what is the problem. But
> first a little question before the "main issue".
>
>
> First - and in order to not create a second thread for a kind of related
> question - I have a little question : would it be possible to create a
> SLiM simulation running forward, output the trees, sample some
> individuals and add mutations afterwards on them and their corresponding
> trees *but with a set number S of SNP across the genomes and the trees*
> First - and in order to not create a second thread for a kind of related
> question - I have a little question : would it be possible to create a
> SLiM simulation running forward, output the trees, sample some
> individuals and add mutations afterwards on them and their corresponding
> i.e, a population of Ne individuals simulated forward during g
> generations of selfing, n individuals are sampled and we want the
> corresponding sequences and genealogies (in regard to the branches
> length) of these n individuals to *only contain a number S of SNP*.
> generations of selfing, n individuals are sampled and we want the
> corresponding sequences and genealogies (in regard to the branches
>
> *- Underestimate Fis - *
>
> *Overall context* : I'm trying to simulate a quite simple population
> with selfing for some generations, sample a few individuals and compute
> the corresponding Fis.
>
> Here are the set of _parameters used for the simulations_ :
> the corresponding Fis.
>
>
> * Ne = 500
> * µ = [1e-5, 1e-6, 1e-7, 1e-8]
> * L = 100 kb
> * r = 1e-6
> * s = [0, 0.1, 0.2, 0.3, ... , 0.9, 1.0]
>
>
> *1) SLiM + recap*
> I first run SLiM simulations for a 2 000 generations, save the .trees
> corresponding files, recap and add mutations afterward with pyslim.
> Then, I use a python script to compute the Fis - so the pipeline is
> something like this : _SLiM -> recap -> mutate -> Fis_.
> corresponding files, recap and add mutations afterward with pyslim.
> Then, I use a python script to compute the Fis - so the pipeline is
>
> However, *the observed Fis were usually a bit lower than expected,
> especially for high value of Fis*.
>
> In order to see if something might have gone wrong with the recapitation
> or the "mutation" process, I then run some fully forward simulations.
> *
> In order to see if something might have gone wrong with the recapitation
> or the "mutation" process, I then run some fully forward simulations.
> *
> *2) Fully forward SLiM*
>
> This time, I first let the simulation run for 10.Ne generations without
> selfing before setting s to the desired value and I let the simulation
> run for about 2000 generations.
> I then output in ms format a sample and compute the corresponding Fis -
> so this time, we've got something like this : _SLiM -> msOutput -> Fis_.
> This time, I first let the simulation run for 10.Ne generations without
> selfing before setting s to the desired value and I let the simulation
> run for about 2000 generations.
> I then output in ms format a sample and compute the corresponding Fis -
>
> The results seems to be a bit better with this approach (which bugs me -
> things should be more or less the same no matter what method I'm using)
> but the *Fis are still too low compared to the expected value*.
> The results seems to be a bit better with this approach (which bugs me -
> things should be more or less the same no matter what method I'm using)
>
> Here's the *SLiM script* :
>
> Fis_too_low.png
>
> /x = expected Fis, y = simulated Fis. Different colors show different
> value of µ. Dots are the average value of Fis. The red line is the
> "perfect" simulated = expected case./
>
> So it seems that a lower mutation rate would be better, but my issue is
> more that I don't understand why I have an underestimation of Fis ? It
> might be an issue with my SLiM script itself that cause this, but having
> the same results with 2 different approaches seems a bit strange to me.
>
> Once again, thanks in advance for your time and your answers,
>
> G. L-F
>
> --
> So it seems that a lower mutation rate would be better, but my issue is
> more that I don't understand why I have an underestimation of Fis ? It
> might be an issue with my SLiM script itself that cause this, but having
> the same results with 2 different approaches seems a bit strange to me.
>
> Once again, thanks in advance for your time and your answers,
>
> G. L-F
>
> SLiM forward genetic simulation: http://messerlab.org/slim/
> <http://messerlab.org/slim/>
> ---
> You received this message because you are subscribed to the Google
> Groups "slim-discuss" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to slim-discuss...@googlegroups.com
> <mailto:slim-discuss...@googlegroups.com>.
> To view this discussion on the web visit
> https://groups.google.com/d/msgid/slim-discuss/c646452e-ef1d-4825-97ce-69c1e0b054e0n%40googlegroups.com
> <https://groups.google.com/d/msgid/slim-discuss/c646452e-ef1d-4825-97ce-69c1e0b054e0n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
Miguel de Navascués
UMR CBGP, INRAE
Centre de Biologie pour la Gestion des Populations
755 avenue du campus Agropolis
CS30016
34988 Montferrier-sur-Lez cedex (France)
phone: +33499623370
fax: +33499623345
e-mail: miguel.navascues AT inrae.fr
Guillaume Lan-Fong
Nov 24, 2021, 10:48:03 AM11/24/21
to slim-discuss
Hi Miguel,
Thanks for you answer ! I
indeed calculate the expected Fis as Fis = s / (2-s) but was thinking
that the equilibrium was supposed to be between mutation and genetic
drift first and then expect the effect of selfing to be "quick enough to
stabilize" if I may say so to be estimated by this fomula.
I've tried to run some simulations using your advices, but the end results still
seem to be off the expected Fis ? I don't really understand what might
cause that - I might have misunderstood your advices so I'll provide you
my SLiM script too.
- SLiM script -
//// A HELPER FUNCTION FOR SETTING CONSTANTS THAT MIGHT BE CONFIGURED VIA COMMAND LINE.
function (void) defineCfgParam(string$ name, lifs value) {
if (!exists(name))
defineConstant(name, value);
}
// set up a simple neutral simulation
initialize() {
//initializeTreeSeq();
defineCfgParam("sim_id", "200");
defineCfgParam("Ne", 500);
defineCfgParam("mu", 1e-8);
defineCfgParam("s", 0.5);
defineCfgParam("samp", 20);
function (void) defineCfgParam(string$ name, lifs value) {
if (!exists(name))
defineConstant(name, value);
}
// set up a simple neutral simulation
initialize() {
//initializeTreeSeq();
defineCfgParam("sim_id", "200");
defineCfgParam("Ne", 500);
defineCfgParam("mu", 1e-8);
defineCfgParam("s", 0.5);
defineCfgParam("samp", 20);
p1.setSelfingRate(s);
sim.rescheduleScriptBlock(s1, start=20*Ne, end=20*Ne);
}
s1 20 late() {
sim.rescheduleScriptBlock(s1, start=20*Ne, end=20*Ne);
}
s1 20 late() {
indGen_samp = p1.sampleIndividuals(samp).genomes;
//sim.treeSeqOutput("../results/" + asString(sim_id) + ".trees");
indGen_samp.outputMS("../results/selfEq/" + asString(sim_id) + ".ms", filterMonomorphic=F);
}
}
#############
I'm using the same parameters as before, here's a reminder :
- Ne = 500
- µ = [1e-5, 1e-6, 1e-7, 1e-8]
- L = 100 kb
- r = 1e-6
- s = [0, 0.1, 0.2, 0.3, ... , 0.9, 1.0]
And here are the results of these simulations :
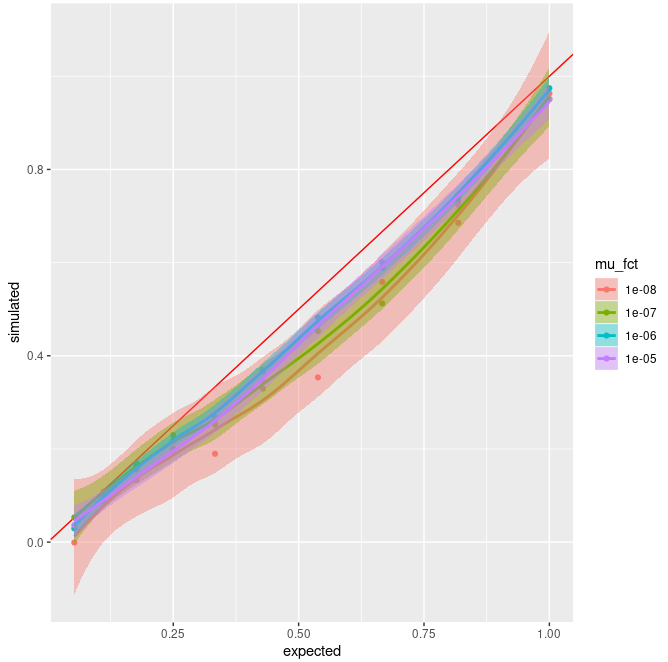
I
do find it bugging that I don't observe random fluctuations but a clear
underestimation in both cases. That's why I was trying to get a way to
simulate a very simple and "easy to tweak and check manually" case using
a set number of SNP through the all tree (thus my first "little"
question).
Thanks for you help anyway !
G. L-F
Miguel de Navascués
Nov 24, 2021, 4:37:44 PM11/24/21
to slim-discuss
Hi Guillaume,
It might not be very intuitive but Fis does not get quickly to the
equilibrium state. If you are pursuing this verification, I would
suggest some additional things to make sure you are getting the expected
results:
* Verify the calculation in your estimation of Fis. Which estimator are
you using? Is it unbiased (e.g. Weir and Cockerham)? I strongly suspect
this could be the issue. You might also want to use independent loci
(one per chromosome, for instance) and increase the sample size (you
could use all individuals).
* Output data and calculate Fis every generation (or every few
generations) and verify its progress through time. Does it look that it
gets closer to the asymptotic value of s/(2-s) or does it stall at a
lower value?
* Make sure that you are at equilibrium in a more rigorous way than just
running 20N generations. First simulate until coalescence has been
achieved (activate tree sequencing recording in SLiM for this) then
simulate for further 10N generations.
Best,
Miguel
On 24/11/2021 16:48, Guillaume Lan-Fong wrote:
>
> Hi Miguel,
>
> Thanks for you answer ! I indeed calculate the expected Fis as Fis = s /
> (2-s) but was thinking that the equilibrium was supposed to be between
> mutation and genetic drift first and then expect the effect of selfing
> to be "quick enough to stabilize" if I may say so to be estimated by
> this fomula.
>
> I've tried to run some simulations using your advices, but the end
> results still seem to be off the expected Fis ? I don't really
> understand what might cause that - I might have misunderstood your
> advices so I'll provide you my SLiM script too.
> *
> *
> *- SLiM script -*
> *
> *
> *#############*
> <https://groups.google.com/d/msgid/slim-discuss/c646452e-ef1d-4825-97ce-69c1e0b054e0n%40googlegroups.com?utm_medium=email&utm_source=footer
> fax: +33499623345 <tel:+33%204%2099%2062%2033%2045>
> e-mail: miguel.navascues AT inrae.fr <http://inrae.fr>
> <https://groups.google.com/d/msgid/slim-discuss/ffb72762-63f6-4a36-8992-be0d07f5ec45n%40googlegroups.com?utm_medium=email&utm_source=footer>.
It might not be very intuitive but Fis does not get quickly to the
equilibrium state. If you are pursuing this verification, I would
suggest some additional things to make sure you are getting the expected
results:
* Verify the calculation in your estimation of Fis. Which estimator are
you using? Is it unbiased (e.g. Weir and Cockerham)? I strongly suspect
this could be the issue. You might also want to use independent loci
(one per chromosome, for instance) and increase the sample size (you
could use all individuals).
* Output data and calculate Fis every generation (or every few
generations) and verify its progress through time. Does it look that it
gets closer to the asymptotic value of s/(2-s) or does it stall at a
lower value?
* Make sure that you are at equilibrium in a more rigorous way than just
running 20N generations. First simulate until coalescence has been
achieved (activate tree sequencing recording in SLiM for this) then
simulate for further 10N generations.
Best,
Miguel
On 24/11/2021 16:48, Guillaume Lan-Fong wrote:
>
> Hi Miguel,
>
> Thanks for you answer ! I indeed calculate the expected Fis as Fis = s /
> (2-s) but was thinking that the equilibrium was supposed to be between
> mutation and genetic drift first and then expect the effect of selfing
> to be "quick enough to stabilize" if I may say so to be estimated by
> this fomula.
>
> I've tried to run some simulations using your advices, but the end
> results still seem to be off the expected Fis ? I don't really
> understand what might cause that - I might have misunderstood your
> advices so I'll provide you my SLiM script too.
> *
> *- SLiM script -*
> *
> *#############*
>
> I'm using the same parameters as before, here's a reminder :
>
> I'm using the same parameters as before, here's a reminder :
>
> * Ne = 500
> * µ = [1e-5, 1e-6, 1e-7, 1e-8]
> * L = 100 kb
> * r = 1e-6
> * s = [0, 0.1, 0.2, 0.3, ... , 0.9, 1.0]
>
>
> * µ = [1e-5, 1e-6, 1e-7, 1e-8]
> * L = 100 kb
> * r = 1e-6
> * s = [0, 0.1, 0.2, 0.3, ... , 0.9, 1.0]
>
>
> And here are the results of these simulations :
>
>
> <https://groups.google.com/d/msgid/slim-discuss/c646452e-ef1d-4825-97ce-69c1e0b054e0n%40googlegroups.com?utm_medium=email&utm_source=footer>>.
>
>
>
> --
> Miguel de Navascués
>
> UMR CBGP, INRAE
> Centre de Biologie pour la Gestion des Populations
> 755 avenue du campus Agropolis
> CS30016
> 34988 Montferrier-sur-Lez cedex (France)
>
> phone: +33499623370 <tel:+33%204%2099%2062%2033%2070>
>
>
>
> --
> Miguel de Navascués
>
> UMR CBGP, INRAE
> Centre de Biologie pour la Gestion des Populations
> 755 avenue du campus Agropolis
> CS30016
> 34988 Montferrier-sur-Lez cedex (France)
>
> fax: +33499623345 <tel:+33%204%2099%2062%2033%2045>
> e-mail: miguel.navascues AT inrae.fr <http://inrae.fr>
>
> --
> SLiM forward genetic simulation: http://messerlab.org/slim/
> <http://messerlab.org/slim/>
> ---
> You received this message because you are subscribed to the Google
> Groups "slim-discuss" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to slim-discuss...@googlegroups.com
> <mailto:slim-discuss...@googlegroups.com>.
> To view this discussion on the web visit
> https://groups.google.com/d/msgid/slim-discuss/ffb72762-63f6-4a36-8992-be0d07f5ec45n%40googlegroups.com
> --
> SLiM forward genetic simulation: http://messerlab.org/slim/
> <http://messerlab.org/slim/>
> ---
> You received this message because you are subscribed to the Google
> Groups "slim-discuss" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to slim-discuss...@googlegroups.com
> <mailto:slim-discuss...@googlegroups.com>.
> To view this discussion on the web visit
> <https://groups.google.com/d/msgid/slim-discuss/ffb72762-63f6-4a36-8992-be0d07f5ec45n%40googlegroups.com?utm_medium=email&utm_source=footer>.
Reply all
Reply to author
Forward
0 new messages