Thank you very much for taking the time to address my long set of questions. Following up:
1. My (novice) interpretation of the QQ plots I attached to the last message was that the residuals were not normal with the raw data (suggested by their deviation from a line with a slope of 1), and that the ordered quantile normalization made there residuals more normal. Based on my QQ plots, are you not concerned about the normality of the residuals of the raw data for use in a multiple QTL model?
That a large effect QTL (such as the one on chr3, and even on chr2) can make the data bimodal makes perfect sense.
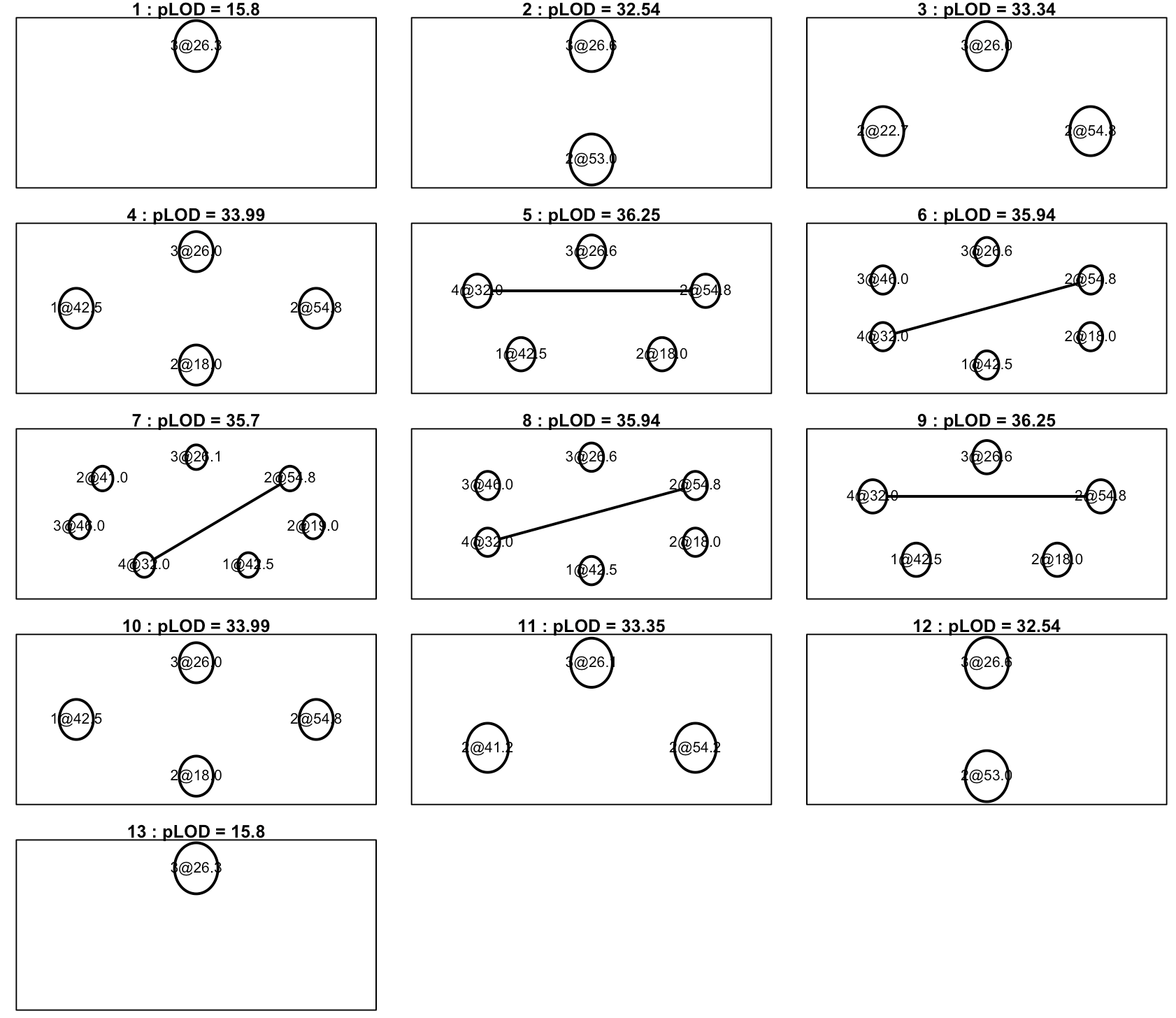
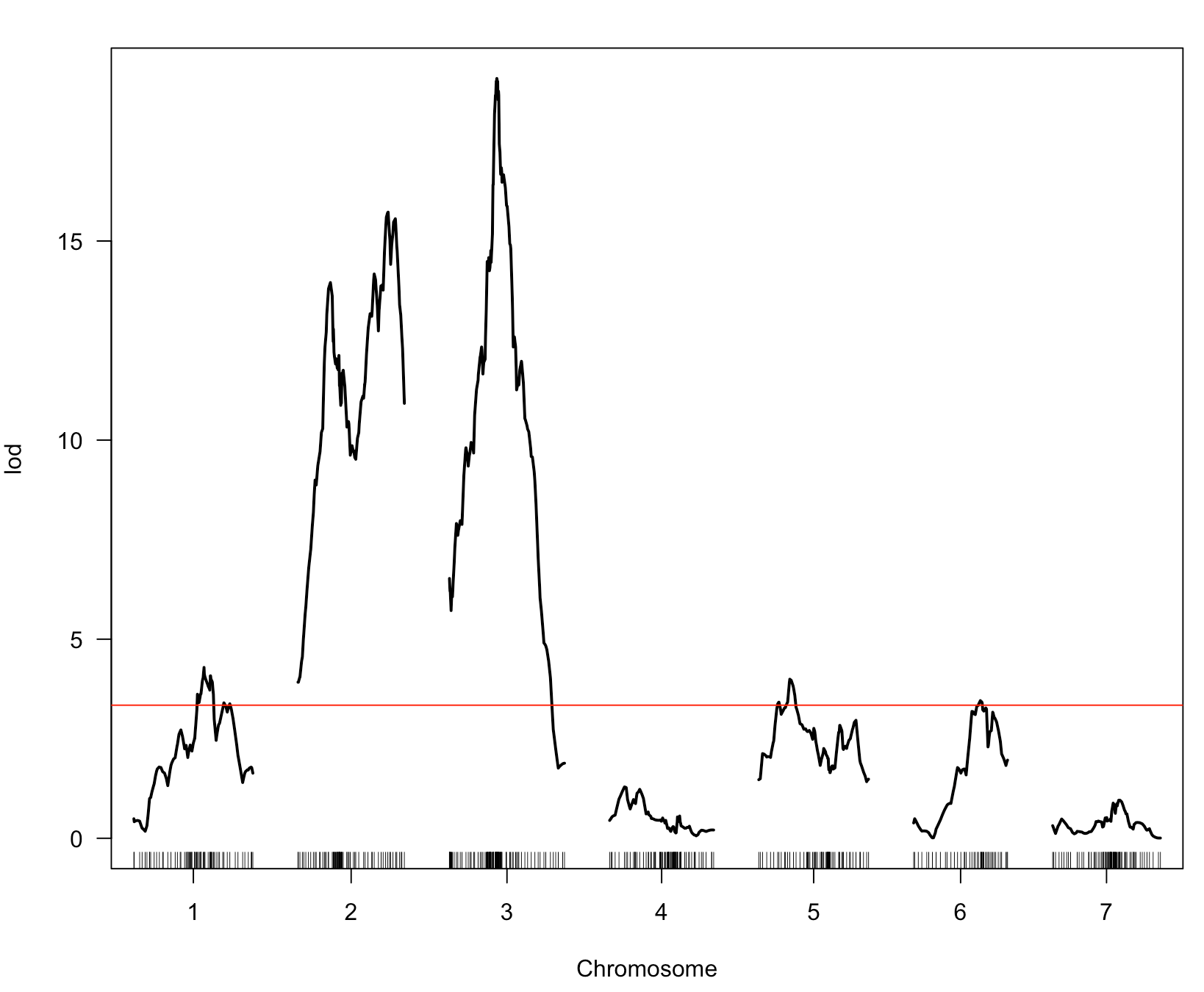
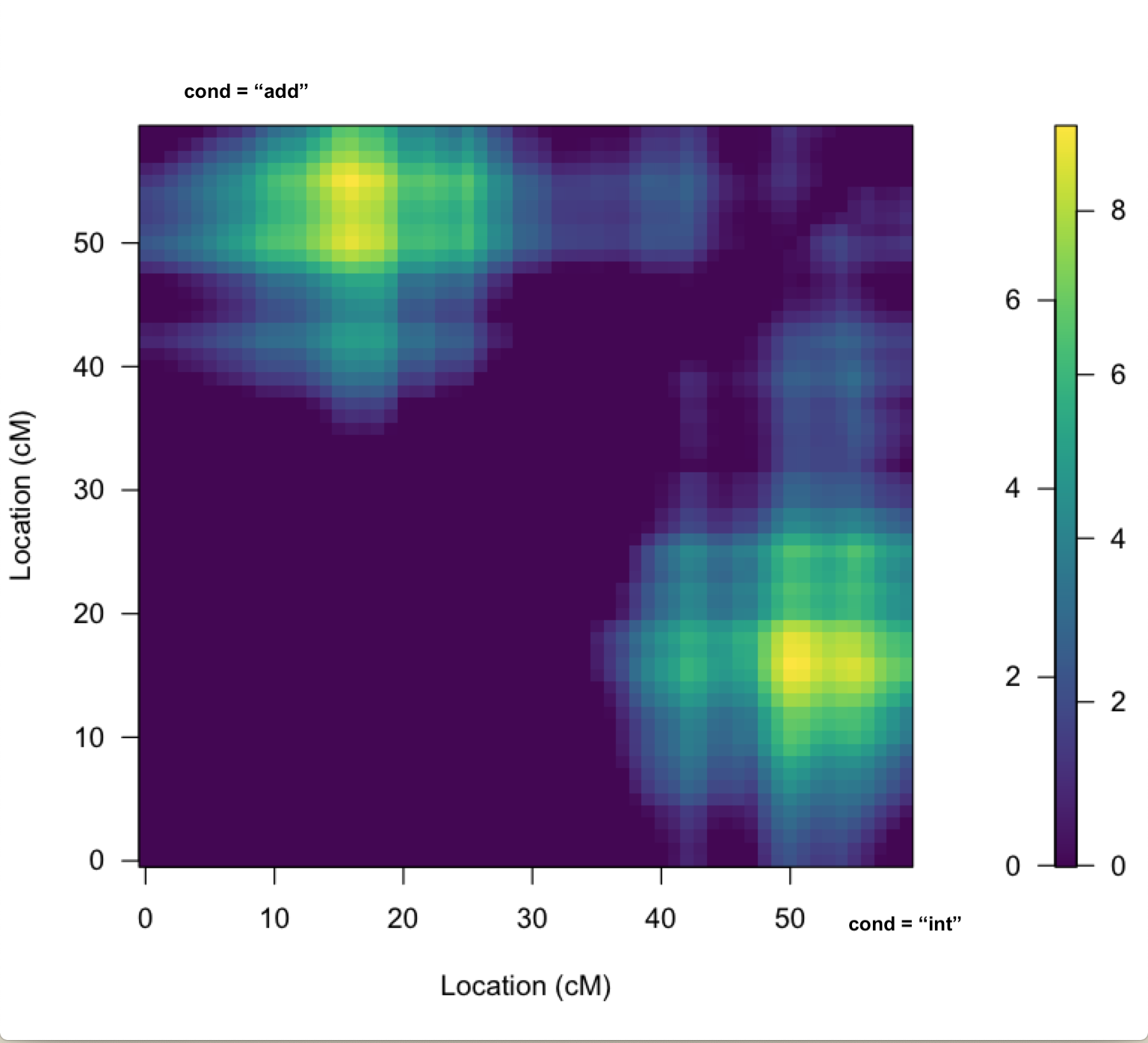
2. There appear to be 3 QTL on chr 2, per the results of scanone and scantwo (attached to previous message), as well as a heat map plot (attached to this message): 1 between 10 & 20cM (A), 1 between 40 & 45cM (B), & 1 around 50cM (C). B & C could certainly be driven by the same gene(s) per the dynamics of imperfect genotyping and our imperfect physical map. I don't expect to work that out with this analysis. However, it does seem like QTL A is worth including in the final model.
The problem is that the multiple QTL analysis, per 'refineqtl' does not include locus A until I consider 4 unique QTL on chr2, which seems excessive given the proximity of the loci it retains before including locus A. Specifically, when 2 QTL are considered (I input 16 & 50 per the results of scantwo: mod1) it refines the loci to 41 & 55cM. This result is yielded with both Haley-Knott and multiple imputation (n.draws = 1000) models. This seems reasonable given the scanone plot. However, when I include 3 loci (both with 'hk' & 'imp': mod2) the loci are refined to 40, 41, & 55cM. Only when I add a 4th chr2 locus does it include a QTL at 14.6cM (mod3).
For my final model, would you recommend just using 'makeqtl' to manually build a model, forcing the 3 chr2 loci to 14.6, 40, & 55cM, and keep all of the refined loci on other chromosomes the same, as I've done in mod4? Or is there another best practices method for dealign with this that I missed?
The code and output associated with my above explanation are included below. It's a lot of text, but I thought it might be helpful for you to reference. Thank you very much for any additional guidance you can provide.
> std_210824 <- sim.geno(std_210824, step = 1, err = 0.01, n.draws = 1000)
> std_210824 <- calc.genoprob(std_210824, step=1, error.prob=0.01)
>
> # model 1: 2 on 2, 2 int on 5
> #hk method
> mod1_hk <- makeqtl(std_210824, chr=c(1,2,2,3,5,5,6), pos = c(38.9, 16, 50, 26, 23, 40, 45), what = "prob")
> mod1_hk_refined <- refineqtl(std_210824, qtl=mod1_hk, formula = y~Q1+Q2+Q3+Q4+Q5*Q6+Q7, verbose = FALSE, pheno.col = "std", method = "hk")
> mod1_hk_refined
QTL object containing genotype probabilities.
name chr pos n.gen
Q1 1@42.5 1 42.478 3
Q2 2@41.2 2 41.179 3
Q3 2@54.8 2 54.819 3
Q4 3@26.3 3 26.269 3
Q5 5@31.0 5 31.000 3
Q6 5@46.1 5 46.144 3
Q7 6@44.7 6 44.682 3
>
> mod1_hk_fit <- fitqtl(std_210824, qtl=mod1_hk_refined, formula = y~Q1+Q2+Q3+Q4+Q5*Q6+Q7, dropone = TRUE, pheno.col = "std", method = "hk")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod1_hk_fit)
fitqtl summary
Method: Haley-Knott regression
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q5:Q6
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 18 1236.6236 68.701313 56.69334 69.31416 0 0
Error 202 547.4616 2.710206
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.5 2 47.96 4.030 2.688 8.848 0.000 0.000207 ***
2@41.2 2 106.36 8.520 5.962 19.622 0.000 1.63e-08 ***
2@54.8 2 39.84 3.371 2.233 7.350 0.000 0.000829 ***
3@26.3 2 397.59 26.200 22.285 73.351 0.000 < 2e-16 ***
5@31.0 6 72.58 5.974 4.068 4.463 0.000 0.000289 ***
5@46.1 6 56.35 4.701 3.158 3.465 0.001 0.002788 **
6@44.7 2 38.94 3.298 2.183 7.185 0.001 0.000968 ***
5@31.0:5@46.1 4 45.35 3.819 2.542 4.183 0.001 0.002819 **
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>
> #imp method
> mod1_imp <- makeqtl(std_210824, chr=c(1,2,2,3,5,5,6), pos = c(38.9, 16, 50, 26, 23, 40, 45))
> mod1_imp_refined <- refineqtl(std_210824, qtl=mod1_imp, formula = y~Q1+Q2+Q3+Q4+Q5*Q6+Q7, verbose = FALSE, pheno.col = "std")
> mod1_imp_refined
QTL object containing imputed genotypes, with 1000 imputations.
name chr pos n.gen
Q1 1@42.3 1 42.275 3
Q2 2@37.2 2 37.236 3
Q3 2@40.9 2 40.921 3
Q4 3@26.7 3 26.721 3
Q5 5@8.3 5 8.316 3
Q6 5@44.3 5 44.316 3
Q7 6@44.7 6 44.682 3
>
> mod1_imp_fit <- fitqtl(std_210824, qtl=mod1_imp_refined, formula = y~Q1+Q2+Q3+Q4+Q5*Q6+Q7, dropone = TRUE, pheno.col = "std")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod1_imp_fit)
fitqtl summary
Method: multiple imputation
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q5:Q6
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 18 1288.3055 71.572525 61.45201 72.21098 0 0
Error 202 495.7798 2.454355
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.3 2 46.36 4.290 2.599 9.445 0.000 0.000120 ***
2@37.2 2 39.21 3.653 2.198 7.988 0.000 0.000458 ***
2@40.9 2 125.27 10.811 7.022 25.521 0.000 1.31e-10 ***
3@26.7 2 441.20 30.547 24.730 89.881 0.000 < 2e-16 ***
5@8.3 6 96.55 8.539 5.411 6.556 0.000 2.42e-06 ***
5@44.3 6 83.77 7.492 4.695 5.689 0.000 1.75e-05 ***
6@44.7 2 31.22 2.931 1.750 6.361 0.001 0.002095 **
5@8.3:5@44.3 4 67.61 6.135 3.790 6.887 0.000 3.24e-05 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>
>
> # model 2: 3 on chr2, 2 int on chr 5
> #hk
> mod2_hk <- makeqtl(std_210824, chr=c(1,2,2,2,3,5,5,6), pos = c(38.9, 16, 41, 50, 26, 23, 40, 45), what = "prob")
> mod2_hk_refined <- refineqtl(std_210824, qtl=mod2_hk, formula = y~Q1+Q2+Q3+Q4+Q5+Q6*Q7+Q8, verbose = FALSE, pheno.col = "std", method = "hk")
> mod2_hk_refined
QTL object containing genotype probabilities.
name chr pos n.gen
Q1 1@42.5 1 42.478 3
Q2 2@40.0 2 40.000 3
Q3 2@41.2 2 41.179 3
Q4 2@54.8 2 54.819 3
Q5 3@26.3 3 26.269 3
Q6 5@30.5 5 30.540 3
Q7 5@46.1 5 46.144 3
Q8 6@52.1 6 52.074 3
>
> mod2_hk_fit <- fitqtl(std_210824, qtl=mod2_hk_refined, formula = y~Q1+Q2+Q3+Q4+Q5+Q6*Q7+Q8, dropone = TRUE, pheno.col = "std", method = "hk")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod2_hk_fit)
fitqtl summary
Method: Haley-Knott regression
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q8 + Q6:Q7
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 20 1266.9046 63.345228 59.42395 71.01144 0 0
Error 200 517.1807 2.585903
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.5 2 42.75 3.811 2.396 8.266 0.000 0.000355 ***
2@40.0 2 34.07 3.062 1.910 6.588 0.001 0.001695 **
2@41.2 2 42.10 3.755 2.360 8.140 0.000 0.000400 ***
2@54.8 2 39.87 3.564 2.235 7.709 0.000 0.000596 ***
3@26.3 2 401.84 27.590 22.523 77.697 0.000 < 2e-16 ***
5@30.5 6 70.13 6.102 3.931 4.520 0.000 0.000255 ***
5@46.1 6 52.85 4.669 2.962 3.406 0.001 0.003194 **
6@52.1 2 40.03 3.577 2.244 7.739 0.000 0.000579 ***
5@30.5:5@46.1 4 42.03 3.750 2.356 4.064 0.002 0.003441 **
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>
> #imp
> mod2_imp <- makeqtl(std_210824, chr=c(1,2,2,2,3,5,5,6), pos = c(38.9, 16, 41, 50, 26, 23, 40, 45))
> mod2_imp_refined <- refineqtl(std_210824, qtl=mod2_imp, formula = y~Q1+Q2+Q3+Q4+Q5+Q6*Q7+Q8, verbose = FALSE, pheno.col = "std")
> mod2_imp_refined
QTL object containing imputed genotypes, with 1000 imputations.
name chr pos n.gen
Q1 1@42.5 1 42.478 3
Q2 2@38.8 2 38.813 3
Q3 2@41.2 2 41.179 3
Q4 2@54.8 2 54.819 3
Q5 3@26.7 3 26.721 3
Q6 5@30.5 5 30.540 3
Q7 5@46.1 5 46.144 3
Q8 6@51.7 6 51.747 3
>
> mod2_imp_fit <- fitqtl(std_210824, qtl=mod2_imp_refined, formula = y~Q1+Q2+Q3+Q4+Q5+Q6*Q7+Q8, dropone = TRUE, pheno.col = "std")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod2_imp_fit)
fitqtl summary
Method: multiple imputation
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q8 + Q6:Q7
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 20 1309.9455 65.497275 63.59377 73.42393 0 0
Error 200 474.1397 2.370699
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.5 2 44.32 4.289 2.484 9.348 0.000 0.000131 ***
2@38.8 2 31.20 3.059 1.749 6.581 0.001 0.001706 **
2@41.2 2 52.60 5.049 2.948 11.094 0.000 2.70e-05 ***
2@54.8 2 46.19 4.461 2.589 9.743 0.000 9.17e-05 ***
3@26.7 2 426.01 30.764 23.878 89.849 0.000 < 2e-16 ***
5@30.5 6 81.86 7.643 4.588 5.755 0.000 1.52e-05 ***
5@46.1 6 60.90 5.799 3.413 4.281 0.000 0.000440 ***
6@51.7 2 47.19 4.553 2.645 9.952 0.000 7.58e-05 ***
5@30.5:5@46.1 4 50.24 4.833 2.816 5.298 0.000 0.000448 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>
> mod2_refined
QTL object containing imputed genotypes, with 1000 imputations.
name chr pos n.gen
Q1 1@42.5 1 42.478 3
Q2 2@39.0 2 39.000 3
Q3 2@41.2 2 41.179 3
Q4 2@54.8 2 54.819 3
Q5 3@26.0 3 25.969 3
Q6 5@30.5 5 30.540 3
Q7 5@46.1 5 46.144 3
Q8 6@51.7 6 51.747 3
> # puts 2 QTL right next to each other (38 & 41). I wish it wouldn't do this
>
> # LOD = 63, var = 73.2
> # LOD doesn't increase enough to justify 3rd locus on chr2
>
> # model 3: 4 on chr2, 2 int on chr 5
> #hk
> mod3_hk <- makeqtl(std_210824, chr=c(1,2,2,2,2,3,5,5,6), pos = c(38.9, 16, 39, 41, 50, 26, 23, 40, 45), what = "prob")
> mod3_hk_refined <- refineqtl(std_210824, qtl=mod3_hk, formula = y~Q1+Q2+Q3+Q4+Q5+Q6+Q7*Q8+Q9, verbose = FALSE, pheno.col = "std", method = "hk")
> mod3_hk_refined
QTL object containing genotype probabilities.
name chr pos n.gen
Q1 1@42.3 1 42.275 3
Q2 2@14.6 2 14.623 3
Q3 2@40.0 2 40.000 3
Q4 2@41.2 2 41.179 3
Q5 2@54.8 2 54.819 3
Q6 3@26.3 3 26.269 3
Q7 5@32.0 5 32.000 3
Q8 5@39.6 5 39.615 3
Q9 6@44.7 6 44.682 3
>
> mod3_hk_fit <- fitqtl(std_210824, qtl=mod3_hk_refined, formula = y~Q1+Q2+Q3+Q4+Q5+Q6+Q7*Q8+Q9, dropone = TRUE, pheno.col = "std", method = "hk")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod3_hk_fit)
fitqtl summary
Method: Haley-Knott regression
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q8 + Q9 + Q7:Q8
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 22 1282.9492 58.31587 60.93632 71.91076 0 0
Error 198 501.1361 2.53099
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.3 2 51.776 4.7184 2.9021 10.228 0.000 5.92e-05 ***
2@14.6 2 8.247 0.7833 0.4622 1.629 0.165 0.198712
2@40.0 2 24.242 2.2670 1.3588 4.789 0.005 0.009309 **
2@41.2 2 35.977 3.3271 2.0165 7.107 0.000 0.001045 **
2@54.8 2 35.375 3.2733 1.9828 6.988 0.001 0.001168 **
3@26.3 2 324.739 23.9739 18.2020 64.153 0.000 < 2e-16 ***
5@32.0 6 64.710 5.8281 3.6271 4.261 0.000 0.000463 ***
5@39.6 6 59.321 5.3688 3.3250 3.906 0.000 0.001036 **
6@44.7 2 25.752 2.4048 1.4434 5.087 0.004 0.007006 **
5@32.0:5@39.6 4 30.831 2.8651 1.7281 3.045 0.010 0.018262 *
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>
> #imp
> mod3_imp <- makeqtl(std_210824, chr=c(1,2,2,2,2,3,5,5,6), pos = c(38.9, 16, 39, 41, 50, 26, 23, 40, 45))
> mod3_imp_refined <- refineqtl(std_210824, qtl=mod3_imp, formula = y~Q1+Q2+Q3+Q4+Q5+Q6+Q7*Q8+Q9, verbose = FALSE, pheno.col = "std")
> mod3_imp_refined
QTL object containing imputed genotypes, with 1000 imputations.
name chr pos n.gen
Q1 1@42.5 1 42.478 3
Q2 2@13.7 2 13.740 3
Q3 2@38.8 2 38.813 3
Q4 2@41.2 2 41.179 3
Q5 2@54.8 2 54.819 3
Q6 3@26.7 3 26.721 3
Q7 5@30.5 5 30.540 3
Q8 5@46.1 5 46.144 3
Q9 6@51.7 6 51.747 3
>
> mod3_imp_fit <- fitqtl(std_210824, qtl=mod3_imp_refined, formula = y~Q1+Q2+Q3+Q4+Q5+Q6+Q7*Q8+Q9, dropone = TRUE, pheno.col = "std")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod3_imp_fit)
fitqtl summary
Method: multiple imputation
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q8 + Q9 + Q7:Q8
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 22 1318.9614 59.95279 64.5151 73.92929 0 0
Error 198 465.1238 2.34911
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.5 2 48.056 4.7185 2.6936 10.229 0.000 5.92e-05 ***
2@13.7 2 9.016 0.9213 0.5054 1.919 0.120 0.149471
2@38.8 2 21.394 2.1581 1.1992 4.554 0.007 0.011655 *
2@41.2 2 46.091 4.5343 2.5834 9.810 0.000 8.66e-05 ***
2@54.8 2 48.908 4.7981 2.7414 10.410 0.000 5.03e-05 ***
3@26.7 2 355.760 27.2618 19.9408 75.722 0.000 < 2e-16 ***
5@30.5 6 73.243 7.0179 4.1054 5.197 0.000 5.47e-05 ***
5@46.1 6 55.046 5.3677 3.0854 3.905 0.000 0.001039 **
6@51.7 2 42.279 4.1752 2.3698 8.999 0.000 0.000182 ***
5@30.5:5@46.1 4 45.894 4.5159 2.5724 4.884 0.000 0.000890 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>
> # model 4: force the 3 chr2 loci from mod2 to spread out, the first locus assigned per the mod3 refinement
> mod4 <- makeqtl(std_210824, chr=c(1,2,2,2,3,5,5,6), pos = c(42.3, 14.6, 41.2, 55, 26, 1, 44, 47))
> mod4_fit <- fitqtl(std_210824, qtl = mod4, formula = y~Q1+Q2+Q3+Q4+Q5+Q6*Q7+Q8, dropone = TRUE, pheno.col = "std")
Warning message:
In fitqtlengine(pheno = pheno, qtl = qtl, covar = covar, formula = formula, :
Dropping 113 individuals with missing phenotypes.
> summary(mod4_fit)
fitqtl summary
Method: multiple imputation
Model: normal phenotype
Number of observations : 221
Full model result
----------------------------------
Model formula: y ~ Q1 + Q2 + Q3 + Q4 + Q5 + Q6 + Q7 + Q8 + Q6:Q7
df SS MS LOD %var Pvalue(Chi2) Pvalue(F)
Model 20 1302.1966 65.109831 62.81582 72.9896 0 0
Error 200 481.8886 2.409443
Total 220 1784.0852
Drop one QTL at a time ANOVA table:
----------------------------------
df Type III SS LOD %var F value Pvalue(Chi2) Pvalue(F)
1@42.3 2 55.24 5.208 3.096 11.463 0.000 1.94e-05 ***
2@14.6 2 31.45 3.034 1.763 6.526 0.001 0.001797 **
2@41.2 2 72.49 6.725 4.063 15.043 0.000 8.20e-07 ***
2@55.0 2 39.75 3.804 2.228 8.249 0.000 0.000361 ***
3@26.0 2 282.14 22.118 15.814 58.549 0.000 < 2e-16 ***
5@1.0 6 75.26 6.964 4.218 5.206 0.000 5.32e-05 ***
5@44.0 6 60.40 5.667 3.386 4.178 0.000 0.000556 ***
6@47.0 2 18.38 1.796 1.030 3.814 0.016 0.023686 *
5@1.0:5@44.0 4 49.35 4.679 2.766 5.120 0.000 0.000600 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
>


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}