Reproducibility of alt/ref counts w/STAR alignment (through RSEM)
129 views
Skip to first unread message

Andreas
Sep 10, 2020, 9:38:10 AM9/10/20
to rna-star
Dear Alex and others,
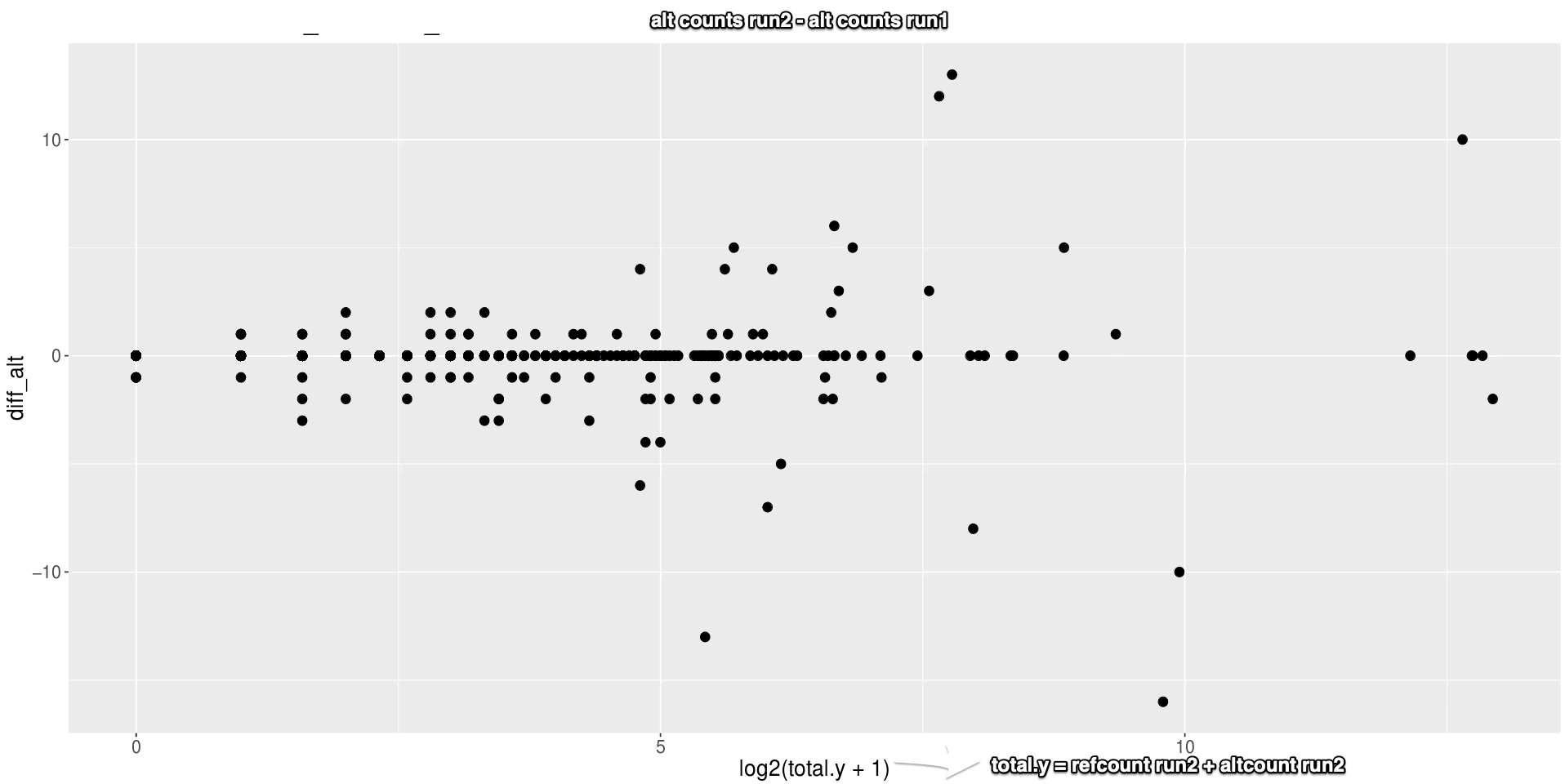
We are using RNA-seq reads to calculate allele-specific expression (+ quantify gene/isoform expression). We are using RSEM (rsem-calculate-expression) with STAR alignment and outputting a genome aligned .bam file (output-genome-gam, rsem).
Then from the genome aligned (and sorted) bam file, we are using a custom pysam pileup script to count reference and alternative alleles based on a input vcf (containing variants).
However, when running rsem/star multiple times (identical settings), the results are not fully reproducible. The bam files are not identical and the count results are not identical (see plot of difference between alt counts for consecutive runs)..
I suspect that this has to do with the fact that STAR is not fully deterministic in the way multimapping reads are output as primary/secondary alignments and the order of them (as described in the STAR manual under multimappers). Also, since we are running multiple threads, setting the seed does also not seem like a valid solution.
Do you have any suggestions on how to make this process deterministic (each run is identical)?
We are running Rsem v1.3.0 & STAR v.2.5.3a
Best Regards,
Andreas Hoff

Alexander Dobin
Sep 17, 2020, 11:57:08 AM9/17/20
to rna-star
Hi Andreas,
we have run into this problem when analyzing data for ENCODE.
STAR is actually deterministic (unless you use --outMultimapperOrder Random) for each read, but with multithreading, the order of the output reads differs from run to run - which affect RSEM quantifications.
The way we solved this was to sort the Aligned.toTranscriptome.out.bam files before feeding them to RSEM.
For PE reads, you would need to sort while keeping the PE reads in pairs, e.g.:
cat <( samtools view -H Tr.bam ) <( samtools view Tr.bam | awk '{printf "%s", $0 "~"; getline; print}' | sort -S 30G -T ./ | tr '~' '\n' ) | samtools view -bS - > Sorted.bam
Such a sorted file should not change from run to run, and RSEM results should be exactly the same.
Cheers
Alex
Reply all
Reply to author
Forward
0 new messages