Fusion reads not well reported in Chimeric.out.sam
469 views
Skip to first unread message

Nico
Aug 21, 2013, 9:05:58 AM8/21/13
to rna-...@googlegroups.com
Hi,
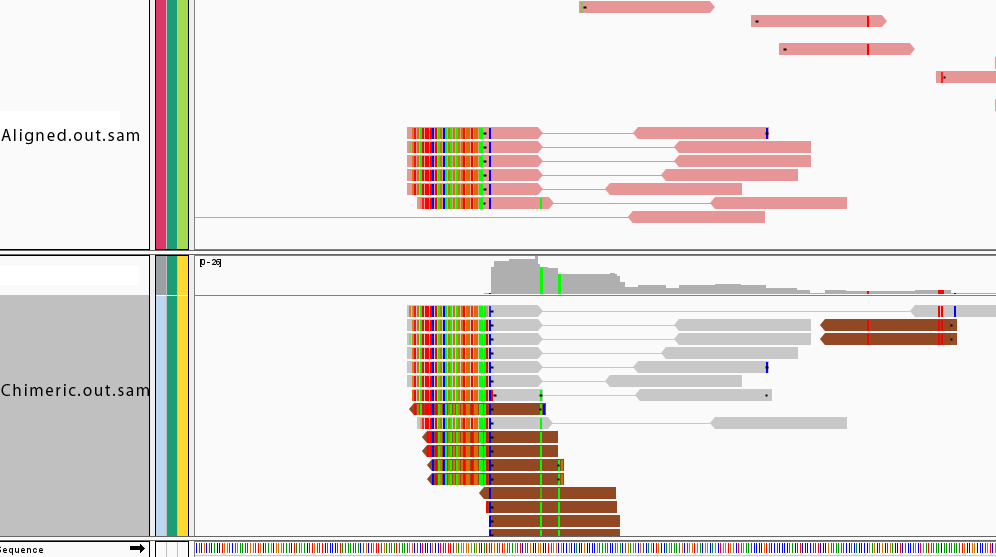
I align my data ( 2x100 stranded) to detect fusion reads. When I check Chimeric.out.sam I found a previously reported fusion. But I found also fusion reads in the Aligned.out.sam file.
Here are two pictures of the data. The top panel is reads from Aligned.out.sam and the bottom one is from Chimeric.out.sam . How can I tune my parameter to have all the fusion reads in Chimeric.out.sam ?
My params :
STAR_2.3.1l/STAR --genomeDir $stargenomeDir --outFilterMultimapNmax 10 --outFilterMismatchNmax 10 --chimSegmentMin 10 --chimJunctionOverhangMin 10 --readFilesIn r1.fastq.gz r2.fastq.gz --readFilesCommand zcat

Thanks a lot
N.

Alexander Dobin
Aug 22, 2013, 11:05:54 AM8/22/13
to rna-...@googlegroups.com
Hi Nico,
each read in Chimeric.out.sam files is supposed to represent only one chimera, i.e. the alignments are unique. For paired-end reads, there will be 2 (for "encompassing" reads) or 3 (for "spanning" reads) lines for each chimeric alignment.
Note, that "spanning" reads might also be reported as non-chimeric in the Aligned.out.sam file. For example, for 2x100b reads, you can have in Chimeric.out.sam one chimeric segment made of 100 read1 bases and 70 read2 bases, and the other chimeric segment made of the remaining 30 read2 bases. The 100+70 bases piece will be reported in Aligned.out.sam file with the 30b soft clipped. I think this would explain your images.
Cheers
Alex
Nico
Aug 23, 2013, 1:41:51 AM8/23/13
to rna-...@googlegroups.com
Thanks for your answer Alex.
But in my example you can see that in Aligned.out.sam, there are also 100+70 bases piece reported while they should be in Chimeric.out.sam.
For the "normale" case, when 100+70 in Chimeric.out.sam and 30 in Aligned.out.sam. Is there a way to remove the 30 segment from Aligned.out.sam and to put it in Chimeric.out.sam ? Maybe by checking the read id ?
Thanks a lot for your help
Alexander Dobin
Aug 23, 2013, 9:19:08 AM8/23/13
to rna-...@googlegroups.com
Hi Nico,
it is possible that STAR finds a linear alignment for 100+70b piece, but cannot align the remaining 30b for some reason (e.g. because of too many mismatches). In that case the 100+70b alignment will be reported in Aligned.out.sam, but there will be no output to Chimeric.out.sam. The 30b segmentcannot be reported in the Aligned.out.sam with your parameters because it's too short. I am not sure how to find the chimeric pairs in your images, I think you need to have read ID printed, or color the reads in different colors.
If you can extract the read of interest from Aligned.out.sam, I can check why the 30b leftover was not mapped chimerically.
Cheers
Alex
Nico
Aug 23, 2013, 9:46:55 AM8/23/13
to rna-...@googlegroups.com
Ok I understand. So the 30bp (soft-clipped) in Aligned.out.sam contain a 1nt gap compared to the reference. Maybe it's that. How can I be less stringent in alignment (mismatch and gap) that the 30bp can align on a other chromosome ( where the fusion point is).
Alexander Dobin
Aug 26, 2013, 7:37:38 PM8/26/13
to rna-...@googlegroups.com
Hi Nico,
I do not think there is a good way to confidently detect a short alignment with an indel in it. You can in principle increase sensitivity of the search (you can try --seedSearchStartLmax 15 --seedSearchLmax 15), but it will also result in a higher rate of false positives, which is more detrimental especially for chimeric detection.
Indels are generally rare in Illumina reads, unless, of course, you have a genomic indel.
Cheers
Alex
Reply all
Reply to author
Forward
0 new messages