building genome for saccharomyces cerevisiae

Kieran Mace
Here is my current workflow:
Download genome data from SGD:
http://www.yeastgenome.org/download-data/sequence
Not sure which files exactly to download, but I'm currently trying:
http://downloads.yeastgenome.org/sequence/S288C_reference/genome_releases/S288C_reference_genome_Current_Release.tgz
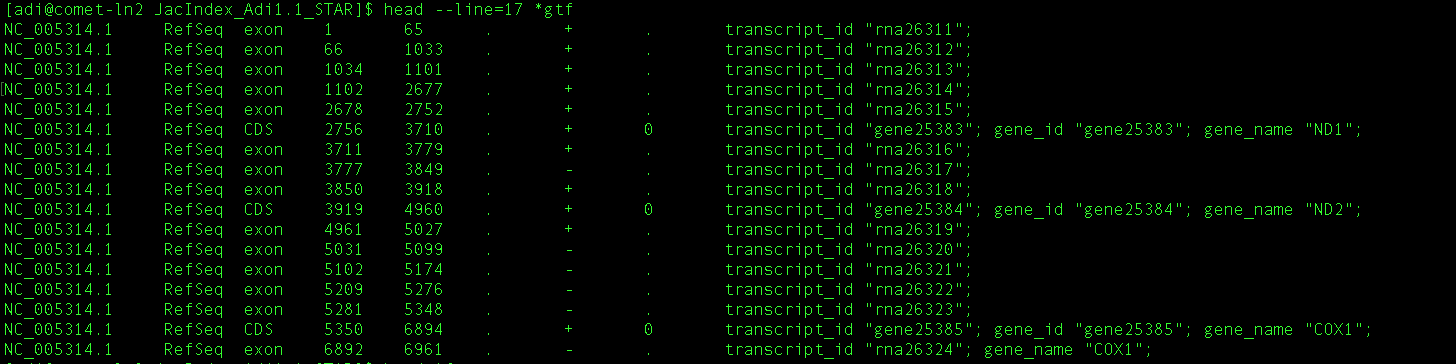
after unzipping this, I then run:
STAR --runThreadN 4 --runMode genomeGenerate --genomeDir genomeDir --genomeFastaFiles S288C_reference_genome_R64-2-1_20150113/S288C_reference_sequence_R64-2-1_20150113.fsa --sjdbGTFfile S288C_reference_genome_R64-2-1_20150113/saccharomyces_cerevisiae_R64-2-1_20150113.gff --sjdbOverhang 300 --sjdbGTFfeatureExon CDS --sjdbGTFtagExonParentTranscript Parent
and getting the following error:
Fatal INPUT FILE error, no valid exon lines in the GTF file: S288C_reference_genome_R64-2-1_20150113/saccharomyces_cerevisiae_R64-2-1_20150113.gff
Solution: check the formatting of the GTF file. Most likely cause is the difference in chromosome naming between GTF and FASTA file.
May 20 15:49:10 ...... FATAL ERROR, exiting

Alexander Dobin
I generally recommend converting the GFF file into GTF, for instance you can use gffread from Cufflinks package to convert gff to gtf:

Aditya Saxena
Alexander Dobin
Aditya Saxena
NC_005314.1 RefSeq exon 1 65 . + . transcript_id "rna26311";
NC_005314.1 RefSeq exon 66 1033 . + . transcript_id "rna26312";
NC_005314.1 RefSeq exon 1034 1101 . + . transcript_id "rna26313";
NC_005314.1 RefSeq exon 1102 2677 . + . transcript_id "rna26314";
NC_005314.1 RefSeq exon 2678 2752 . + . transcript_id "rna26315";
NC_005314.1 RefSeq CDS 2756 3710 . + 0 transcript_id "gene25383"; gene_id "gene25383"; gene_name "ND1";
NC_005314.1 RefSeq exon 3711 3779 . + . transcript_id "rna26316";
NC_005314.1 RefSeq exon 3777 3849 . - . transcript_id "rna26317";
NC_005314.1 RefSeq exon 3850 3918 . + . transcript_id "rna26318";
NC_005314.1 RefSeq CDS 3919 4960 . + 0 transcript_id "gene25384"; gene_id "gene25384"; gene_name "ND2";
NC_005314.1 RefSeq exon 4961 5027 . + . transcript_id "rna26319";
NC_005314.1 RefSeq exon 5031 5099 . - . transcript_id "rna26320";
NC_005314.1 RefSeq exon 5102 5174 . - . transcript_id "rna26321";
NC_005314.1 RefSeq exon 5209 5276 . - . transcript_id "rna26322";
NC_005314.1 RefSeq exon 5281 5348 . - . transcript_id "rna26323";
NC_005314.1 RefSeq CDS 5350 6894 . + 0 transcript_id "gene25385"; gene_id "gene25385"; gene_name "COX1";
NC_005314.1 RefSeq exon 6892 6961 . - . transcript_id "rna26324"; gene_name "COX1";
NC_005314.1 RefSeq exon 6966 7032 . + . transcript_id "rna26325";
NC_005314.1 RefSeq CDS 7034 7717 . + 0 transcript_id "gene25386"; gene_id "gene25386"; gene_name "COX2";
NC_005314.1 RefSeq exon 7723 7787 . + . transcript_id "rna26326";
My genome.fa has no chromosome names as the genome has only unplaced scaffolds (right?). This how the genome.fa file heads.
>gi|484394521|ref|NW_004504313.1| Jaculus jaculus isolate JJ0015 unplaced genomic scaffold, JacJac1.0 scaffold00001, whole genome shotgun sequence
GTCTGTGAGGAAATGACCTACGAGGAAATTCAGGCCCATTATCCACTTGAGTTCGCCCTACACGACCAGG
AGAAGTACCGTTACTGGTATCCGAAGGGTGAGTCCTATGAGGACCTGGTCCAGCGACTGGAGCCTGTCAT
CTTGGAATTGGAGAGACAGGAGAACATGCTGGTCATGTGCCACCAGGCTGTGATGCGAGGCCACCTGGCA
CACTTCAAAGACAAGGCAGCAGAACAGCTGGCCTACCTCAAGTGTCCCCTTCACACGGTCCTGAAGCTGA
CCCTTGTGGCTTACGGCTGTAAAGTGAAGTCCATATTCTTGAATGTGGCAGCTGTGAATACACACTGAGA
CAGGCTGCAGAATGTAGACATCTCCAGGCCTCCAGAGGAAGCCGTTGTCACAGTCTCTGCTCACCAGTGA [.......]
Thanks much for your help and reply!
cheers
Adi
Alexander Dobin
Aditya Saxena
{kind=link}
{kind=link}
Alexander Dobin
Aditya Saxena
>gi|484394521|ref|NW_004504313.1| Jaculus jaculus isolate JJ0015 unplaced genomic scaffold, JacJac1.0 scaffold00001, whole genome shotgun sequence
GTCTGTGAGGAAATGACCTACGAGGAAATT...........
to simple headers in the genome_renamed.fasta like so,
>NW_004504313.1
GTCTGTGAGGAAATGACCTACGAGGAAATT...........
The genomeGenerate step ran to completion with the genome_renamed.fasta and the genome.gtf (generated from .gff with gffread). I was able to map ~95% of my SR-50 reads to this indexed genome.
Thank you for your advice and help! STAR is awesome :)
cheers
Adi