Monophyletic... or?

jj biodiversity

Alexandros Stamatakis
Which tool are you using to visualize the tree and which RAxML output
file are you using as input for your visualization tool?
There are some known problems with tree viewers and support values that
are described in the following paper:
https://pubmed.ncbi.nlm.nih.gov/28369572/
Alexis
> RAXMLhyal20C.JPG
>
> --
> You received this message because you are subscribed to the Google
> Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to raxml+un...@googlegroups.com
> <mailto:raxml+un...@googlegroups.com>.
> To view this discussion on the web visit
> https://groups.google.com/d/msgid/raxml/4bfcc66c-978d-4905-bb65-d7a806fd62f8n%40googlegroups.com
> <https://groups.google.com/d/msgid/raxml/4bfcc66c-978d-4905-bb65-d7a806fd62f8n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
Alexandros (Alexis) Stamatakis
Research Group Leader, Heidelberg Institute for Theoretical Studies
Full Professor, Dept. of Informatics, Karlsruhe Institute of Technology
Affiliated Scientist, Evolutionary Genetics and Paleogenomics (EGP) lab,
Institute of Molecular Biology and Biotechnology, Foundation for
Research and Technology Hellas
www.exelixis-lab.org

Lucas Czech
Dear Jeong,
to add to this, it might also be that you indeed have a branch with virtually zero branch length there. Judging from the images you posted, there are some (almost-) zero branch lengths within these clades, so I am assuming that you are trying to resolve a phylogeny of very closely related sequences there, such as strains from the same species? It might just be that these three involved clades (including the one at the top of your picture that is not shown) are indeed very close to being trifurcating, so that they appear as zero branch lengths.
Alternatively, it might be an issue with the alignment, such as sequences aligning to different regions of the alignment, so that their respective phylogeny cannot be properly resolved:
Seq1 AAAAAA----
Seq2 AAATAA----
Seq3 ----AACCCC
Seq4 ----AACGCC
Here, Seq1 and Seq2 would be in one clade, and Seq3 and Seq4 in
another, but their respective parent branch that leads to these
four sequences cannot properly resolve between the two clades.
That might be worth checking with some alignment viewer, such as AliView.
Lastly, which RAxML version are you using? We always suggest to switch to RAxML-ng, which is the current and maintained version.
Cheers and so long
Lucas
jj biodiversity
Lucas Czech
Well, in that case, it might just be that these are the maximum
likelihood branch lengths for that tree :-) or is there a reason
why that seems improbable?
To unsubscribe from this group and stop receiving emails from it, send an email to raxml+un...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/raxml/8b9d2cec-d0ce-4f2d-88b7-e0b149a7ea15n%40googlegroups.com.
jj biodiversity
Alexandros Stamatakis
Also to answer a previous question, yes for (m)PTP to consider a bunch
of sequences as stemming from one single species they need to be
monophyletic in the tree.
Alexis
>> Lastly, which RAxML version are you using? We always suggest
>>>>
>>>
>> --
>> You received this message because you are subscribed to the Google
>> Groups "raxml" group.
>> To unsubscribe from this group and stop receiving emails from it,
>> send an email to raxml+un...@googlegroups.com.
>> To view this discussion on the web visit
>> https://groups.google.com/d/msgid/raxml/8b9d2cec-d0ce-4f2d-88b7-e0b149a7ea15n%40googlegroups.com
> --
> You received this message because you are subscribed to the Google
> Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send
> <mailto:raxml+un...@googlegroups.com>.
> To view this discussion on the web visit
> <https://groups.google.com/d/msgid/raxml/e0c6f24b-f735-43be-b7fb-cb07b1551448n%40googlegroups.com?utm_medium=email&utm_source=footer>.
jj biodiversity

Grimm
Alexandros Stamatakis
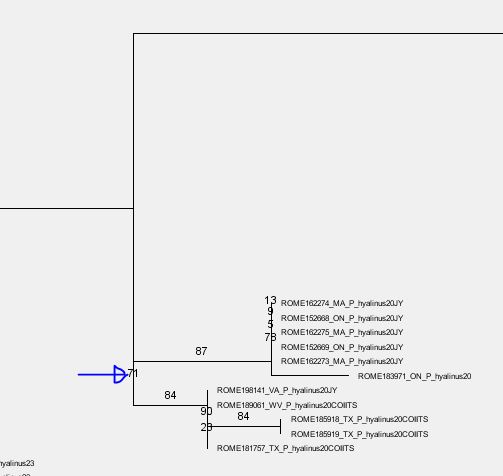
I just visualized it and it seems that the support of 71 you have there
refers to the entire group you are interested in. I assume it might be a
visualization issue due to the short branch lengths.
The partially very small branch lengths are okay here I believe as you
want to delimit species on the tree so you do need/expect near-zero
branch lengths in some parts of the tree.
Alexis
>
> >
> > --
> > You received this message because you are subscribed to the Google
> > Groups "raxml" group.
> > To unsubscribe from this group and stop receiving emails from it,
> send
> > an email to raxml+un...@googlegroups.com
> > <mailto:raxml+un...@googlegroups.com>.
> > To view this discussion on the web visit
> >
> https://groups.google.com/d/msgid/raxml/e0c6f24b-f735-43be-b7fb-cb07b1551448n%40googlegroups.com
> <https://groups.google.com/d/msgid/raxml/e0c6f24b-f735-43be-b7fb-cb07b1551448n%40googlegroups.com>
>
> >
>
>
> --
> Alexandros (Alexis) Stamatakis
>
> Research Group Leader, Heidelberg Institute for Theoretical Studies
> Full Professor, Dept. of Informatics, Karlsruhe Institute of Technology
> Affiliated Scientist, Evolutionary Genetics and Paleogenomics (EGP)
> lab,
> Institute of Molecular Biology and Biotechnology, Foundation for
> Research and Technology Hellas
>
> --
> You received this message because you are subscribed to the Google
> Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to raxml+un...@googlegroups.com
> <mailto:raxml+un...@googlegroups.com>.
> To view this discussion on the web visit
> <https://groups.google.com/d/msgid/raxml/3fba1797-ea89-4afa-8834-8830d1231b51n%40googlegroups.com?utm_medium=email&utm_source=footer>.
jj biodiversity
Grimm
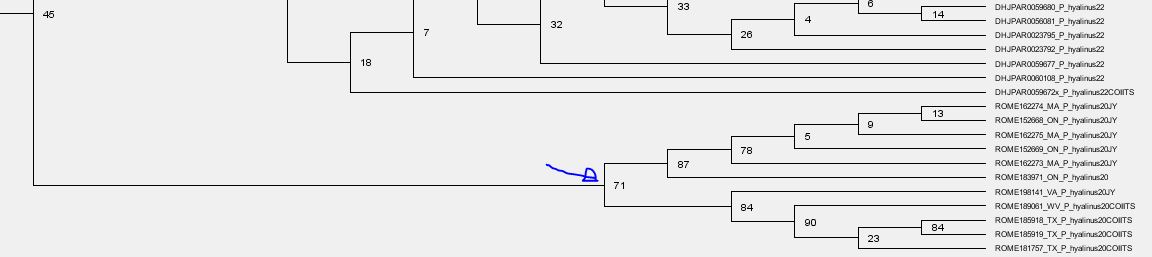
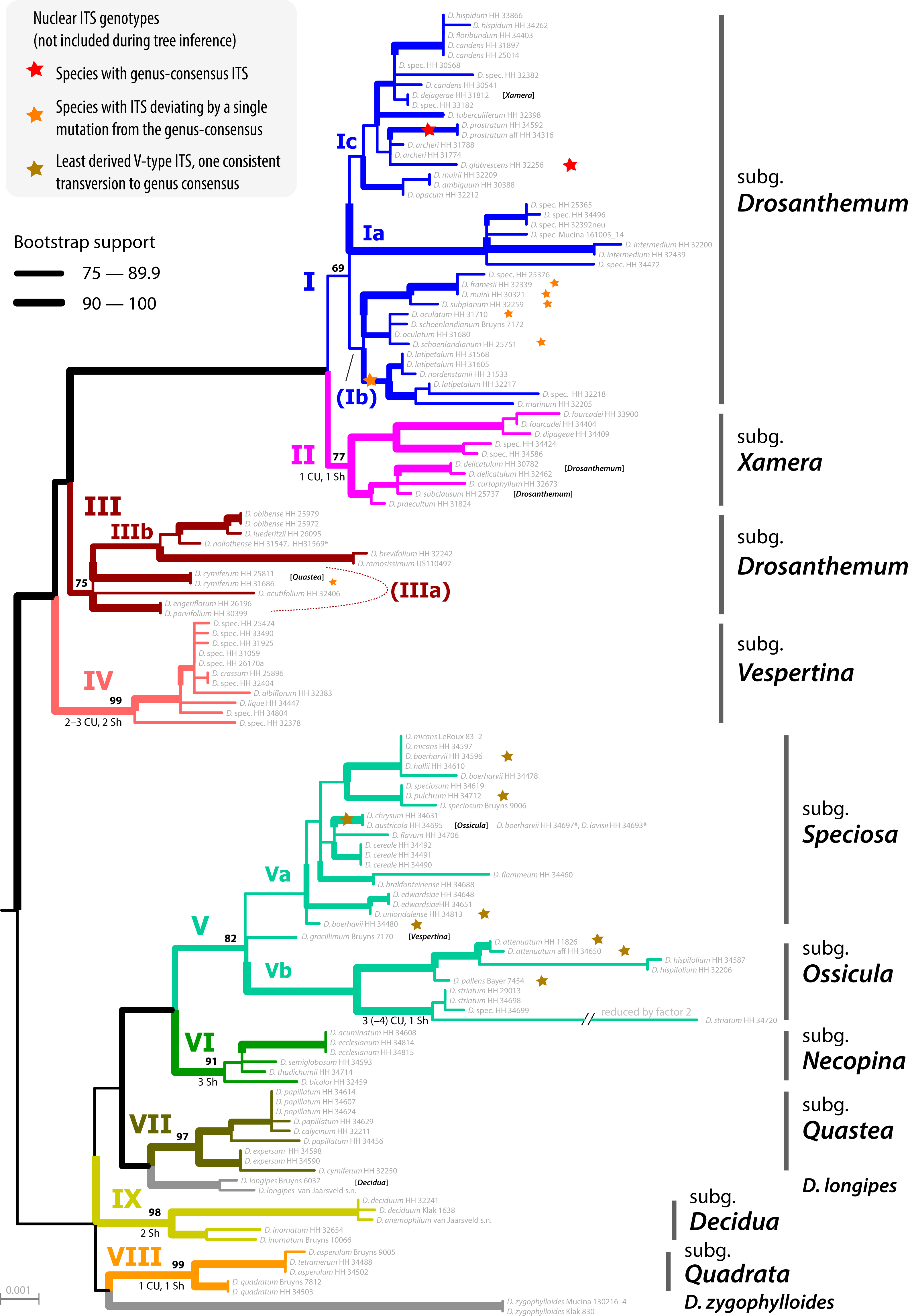
- The next deeper branch, albeit being very prominent, has a BS = 45. Long branches + low BS support are usually related to strongly tree-incompatible signal: we use a tree for data that is not the product of dichotomous evolution. For instance, if you add a chimeric sequence C of two non-sister lineages A and B as "donors", which is 50% A and 50% B, the tree will place it as sister to either A or B with a BS < 50. Your tree may show you BS = 45 for C sister to A, but the BS sample may have equally as much topologies with C sister to B (BS ~ 45). Any mutation patterns shared with the respective other, the rejected sister but donor, conflicting with the tree's branching pattern will inflate the branch leading to C. The same applies if C is a natural recombinant (e.g. virus data, certain fungi) or hybrid of A and B (higher organisms). On the other hand, a low and deep (tree-root proximal) BS support may simply be a long-branching artefact/signal issue because (a) too distant outgroup(s) were used that the tree has to put somewhere but don't really fit anywhere.
- The sister lineage (assuming your tree has the evolutionary correct root) of the ROME species is very distant and (from the screenshots) much more diverse from the two ROME clades. This is a tree topology that may be highly biased by sampling bias. Either because you didn't capture the phylogenetically closer groups of the ROMEs or because they cannot be captured because they are long extinct/ ROMEs split from the rest much earlier than the rest diverged. In such a case the lack of character support for the all-ROME branch indicates that the ROMEs are just relicts of the first radiation which may be monophyletic (sister lineages) or paraphyletic, i.e. independently derived from the last common ancestor of the entire subtree (ROME + long-rooting sister).
- Never only look at the (outgroup-)rooted tree but always also at the actually inferred unrooted tree.
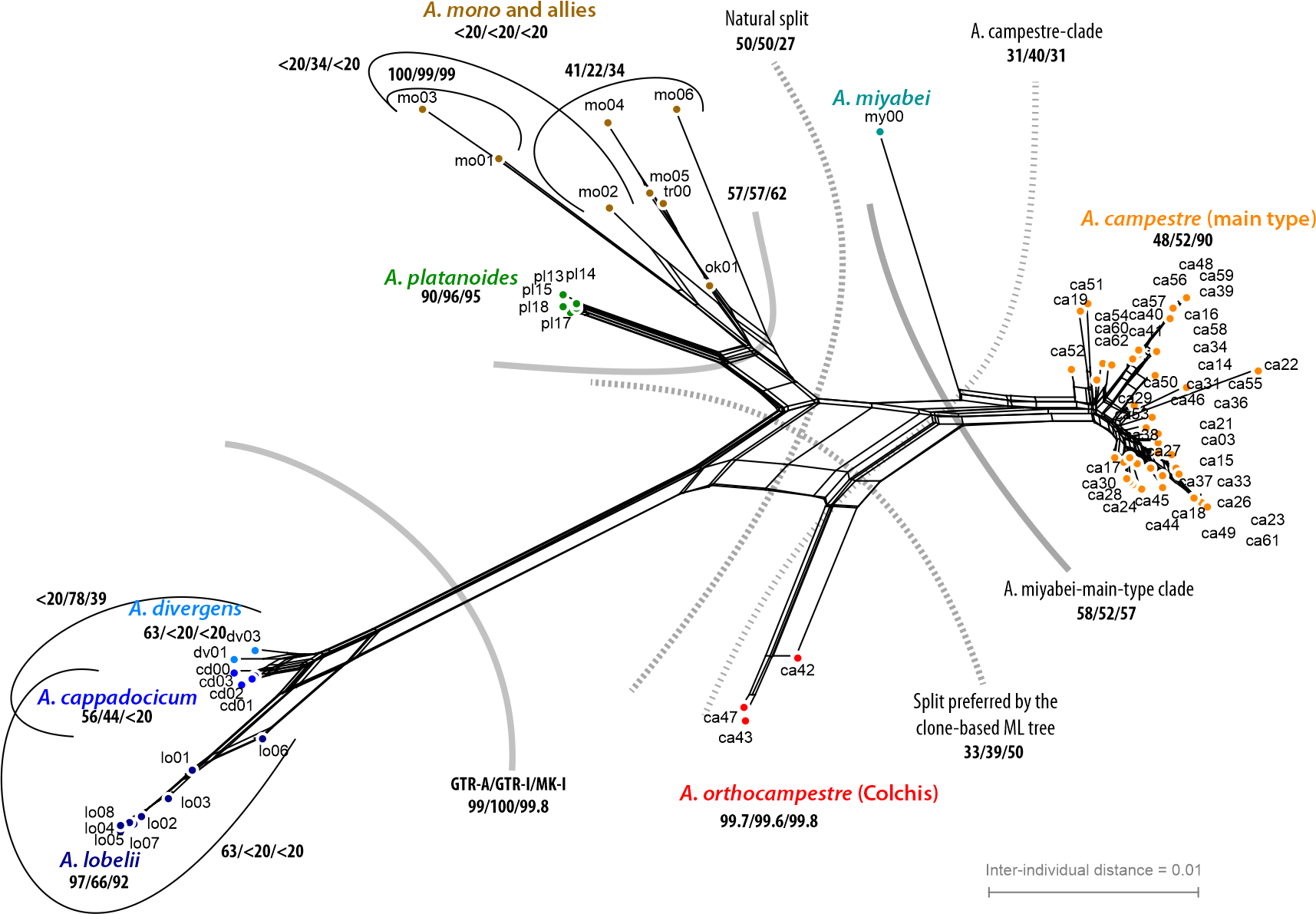
- Check the absolute similarity, the genetic coherence of your clades/putative species: either by phylogeny-sorted (using the inferred tree topology) heat-map or map the clades and eventual ambiguous ML-BS support on a neighbour-net, which is a planar (2-dimensional vs. 1 dimension we model in a tree) distance-based, very quick-to-infer (meta-)phylogenetic network. A further option for easy visualisation of overall diversity to compare with the inferred tree may be phytools new phylogenetic PCoA.
- If you have ambiguous support, use RAxML's bootstrap sample to generate what we called "bootstrap consensus networks". In contrast to a consensus tree, which only mask intra-analysis, intra-data conflict by collapsing non-trivial branches, a consensus network visualises competing topologies.
jj biodiversity
.JPG?part=0.4&view=1)
.JPG?part=0.3&view=1)
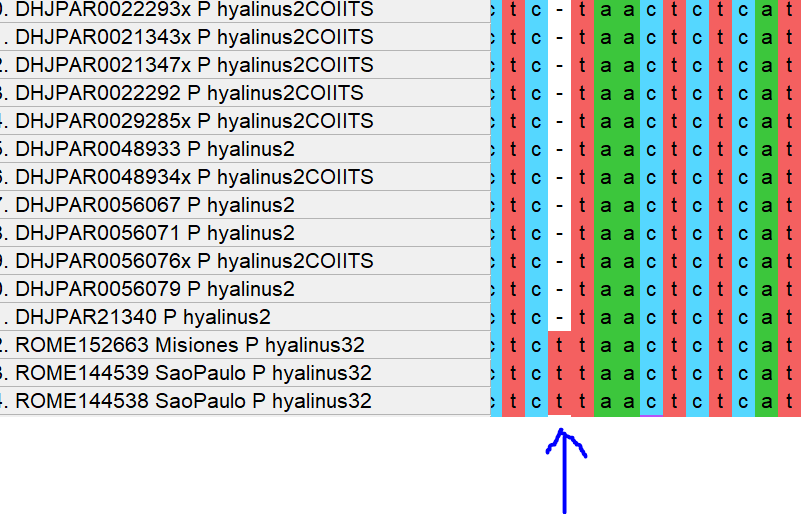
Grimm
-
Being a part of the nuclear-encoded 35S (or 45S) rDNA cistron arrays, they are inherited from both parents. Since these arrays comprise extremely conserved sequence bits (and hundreds if not thousands of them), they are prone to crossing over, so a F1-heterozygote can pass on its paternal, maternal but also recombinant nrDNA arrays. In plants that lack selective fertilisation barriers, especially the wind-pollinated ones, this leads to puzzling intragenomic variation. If you do a direct classic Sange sequencing or NGS like genome skimming you end up with a lot of ambiguous base calls in what is effectively a ITS(2) consensus sequence.
- On the other hand concerted evolution homogenises the arrays, and across the genomes; and this process adds to inbreeding homogenisation effects due to selective fertilisation (e.g. by active choosing of the sex partner, e.g. in birds due to particular courting behaviour). I have not looked at a lot of animal ITS data sets, but this seems to be more or less the rule in most animals. Your ITS(2) sequences are free of ambiguous bases.
- If the 1st+2nd codon-based cox1 tree congruent with the ITS2 tree (or differentiation pattern), the 3rd codon position is biased by saturation effects.
- If the 3rd codon-based cox1 tree is congruent with the ITS2 tree
(or differentiation pattern), the 1st+2nd codon position do not have enough signal to resolve these deep relationships.
jj biodiversity
You received this message because you are subscribed to a topic in the Google Groups "raxml" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/raxml/uOuuXhVC9Q8/unsubscribe.
To unsubscribe from this group and all its topics, send an email to raxml+un...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/raxml/55fea670-98dc-4d9f-a418-e55f22f1606dn%40googlegroups.com.
Grimm
jj biodiversity
Grimm
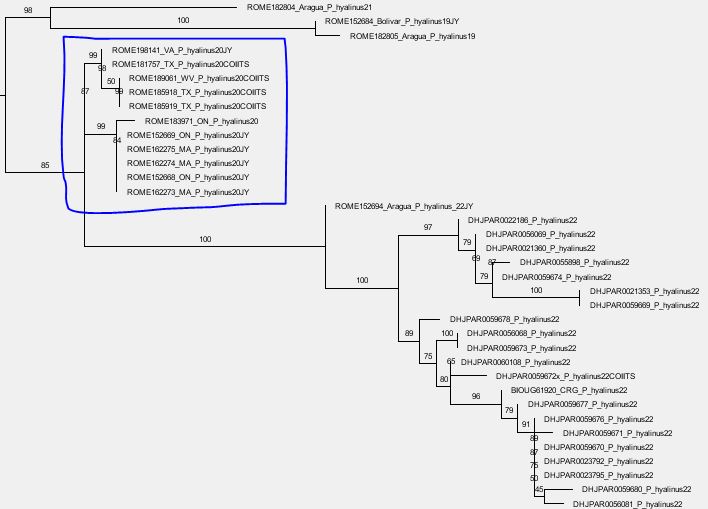
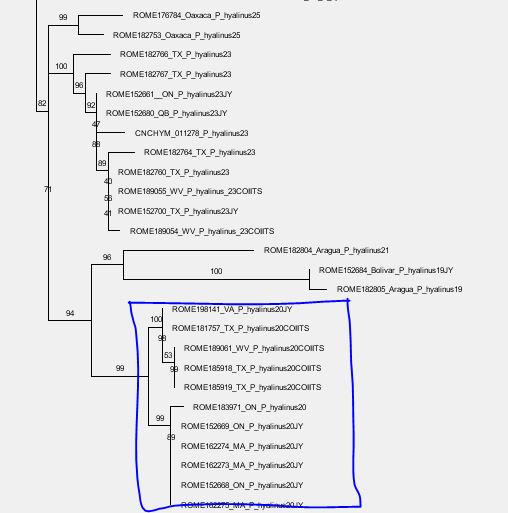
- Standard explanation: they got isolated during the LGM (or an earlier stage), i.e. the maternal differentiation (here: cox1 pattern) happened after the (starting) speciation events (here: ITS2 patterns): few mothers survived, and their slightly different signatures (within-species variation) got established in the according nearby populations.
- Often left-aside alternative: an already mito-polymorphic ancestral species (e.g. a widespread species starting to speciate and bud, with marginal, small populations becoming increasing isolated: differential genetic drift across the total range of the species) was sorted during the Ice Age bottlenecks, with one maternal signature surviving in the one, and the other in another refugium. That is the maternal (cox1) pattern pre-dates the ITS2 differentiation/ongoing speciation.
- Do the cox1 variants of an ITS2 lineage go back to a potentially shared by that ITS2 lineage common cox1 ancestor? This would support scenario 1: Homogenised ancestral species geographically drifting during the Ice Age retractions.
- Or are cox1 variants shared by different, phylogenetically distant ITS2 lineages? This would support scenario 2. Non-homoginised ancestral species being sorted during the Ice Age bootlenecks.