Taxonomy constrained reference 16S tree
50 views
Skip to first unread message

Daniel Lundin
Jun 29, 2021, 10:49:43 AM6/29/21
to ra...@googlegroups.com
Hi,
I'm in the process of creating a reference 16S alignment and
phylogeny, consistent with the GTDB taxonomy. In contrast to GTDB, I'd
prefer a single alignment and phylogeny, i.e. not divided into Archaea
and Bacteria.
It strikes me that the Sativa way of constructing the tree, by
building the phylogeny constrained by a taxonomy tree sounds
promising.
I have a couple of questions (and sorry if they're already answered, I
did search the archive):
1.a I could write a script myself, but I suppose there's code in
Sativa I could use to construct the taxonomy tree? Given a file with a
list of taxon hierarchies like the below or similar, is there a
function I could use to construct the taxonomy tree? (I'm Python
illiterate, but can get help from colleagues if needed.)
"Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Eschericha;;Escherichia
coli"
1.b Or can I abuse Sativa just to output the taxonomy constrained
reference phylo? (I can't see it as a file after a colleague ran
Sativa.)
2. Sativa is documented as using the old RAxML, but I see there's a
--tree-constraint option in RAxML-NG too, so I suppose I can use that?
Does it accept a multifurcating tree?
/Daniel
I'm in the process of creating a reference 16S alignment and
phylogeny, consistent with the GTDB taxonomy. In contrast to GTDB, I'd
prefer a single alignment and phylogeny, i.e. not divided into Archaea
and Bacteria.
It strikes me that the Sativa way of constructing the tree, by
building the phylogeny constrained by a taxonomy tree sounds
promising.
I have a couple of questions (and sorry if they're already answered, I
did search the archive):
1.a I could write a script myself, but I suppose there's code in
Sativa I could use to construct the taxonomy tree? Given a file with a
list of taxon hierarchies like the below or similar, is there a
function I could use to construct the taxonomy tree? (I'm Python
illiterate, but can get help from colleagues if needed.)
"Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Eschericha;;Escherichia
coli"
1.b Or can I abuse Sativa just to output the taxonomy constrained
reference phylo? (I can't see it as a file after a colleague ran
Sativa.)
2. Sativa is documented as using the old RAxML, but I see there's a
--tree-constraint option in RAxML-NG too, so I suppose I can use that?
Does it accept a multifurcating tree?
/Daniel

Lucas Czech
Jun 29, 2021, 3:43:07 PM6/29/21
to ra...@googlegroups.com
Hi Daniel,
we had this issue ourselves before, and instead of "misusing" Sativa, we
have also implemented a proper tool to generate multifurcating trees
from a taxonomy to be used as constraint in tree inference. Please have
a look here: https://github.com/lczech/gappa/wiki/Subcommand:-taxonomy-tree
This should help with your questions 1a and 1b. As for 2, yes, a
multifurcating tree is a way to provide a constraint for tree inference.
If that tree was not multifurcating (that is, if it was fully resolved),
the tree inference would not need to do anything at all, because then
all the branches are resolved already :-D
Hope that helps, and let us know if you have further questions!
Lucas
we had this issue ourselves before, and instead of "misusing" Sativa, we
have also implemented a proper tool to generate multifurcating trees
from a taxonomy to be used as constraint in tree inference. Please have
a look here: https://github.com/lczech/gappa/wiki/Subcommand:-taxonomy-tree
This should help with your questions 1a and 1b. As for 2, yes, a
multifurcating tree is a way to provide a constraint for tree inference.
If that tree was not multifurcating (that is, if it was fully resolved),
the tree inference would not need to do anything at all, because then
all the branches are resolved already :-D
Hope that helps, and let us know if you have further questions!
Lucas

Alexandros Stamatakis
Jun 29, 2021, 11:00:32 PM6/29/21
to ra...@googlegroups.com
Dear Daniel,
There is not much to add to Lucas reply (thanks Lucas :-) ).
Regarding 2: you can and should use the constraint option in RAxML-NG as
a rarely occurring bug in the constrained search option in RAxML was
fixed in RAxML-NG.
All the best,
Alexis
--
Alexandros (Alexis) Stamatakis
Research Group Leader, Heidelberg Institute for Theoretical Studies
Full Professor, Dept. of Informatics, Karlsruhe Institute of Technology
Affiliated Scientist, Evolutionary Genetics and Paleogenomics (EGP) lab,
Institute of Molecular Biology and Biotechnology, Foundation for
Research and Technology Hellas
www.exelixis-lab.org
There is not much to add to Lucas reply (thanks Lucas :-) ).
Regarding 2: you can and should use the constraint option in RAxML-NG as
a rarely occurring bug in the constrained search option in RAxML was
fixed in RAxML-NG.
All the best,
Alexis
Alexandros (Alexis) Stamatakis
Research Group Leader, Heidelberg Institute for Theoretical Studies
Full Professor, Dept. of Informatics, Karlsruhe Institute of Technology
Affiliated Scientist, Evolutionary Genetics and Paleogenomics (EGP) lab,
Institute of Molecular Biology and Biotechnology, Foundation for
Research and Technology Hellas
www.exelixis-lab.org
Daniel Lundin
Jun 30, 2021, 1:27:28 AM6/30/21
to ra...@googlegroups.com
Perfect! Thank you both!
/D
Den ons 30 juni 2021 kl 05:00 skrev Alexandros Stamatakis
<alexandros...@gmail.com>:
> --
> You received this message because you are subscribed to the Google Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send an email to raxml+un...@googlegroups.com.
> To view this discussion on the web visit https://groups.google.com/d/msgid/raxml/54b2e209-bb5a-035e-e29c-bb5f5e8bdad4%40gmail.com.
/D
Den ons 30 juni 2021 kl 05:00 skrev Alexandros Stamatakis
<alexandros...@gmail.com>:
> You received this message because you are subscribed to the Google Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send an email to raxml+un...@googlegroups.com.
> To view this discussion on the web visit https://groups.google.com/d/msgid/raxml/54b2e209-bb5a-035e-e29c-bb5f5e8bdad4%40gmail.com.
Daniel Lundin
Jul 1, 2021, 8:05:49 AM7/1/21
to ra...@googlegroups.com
Hi again,
As a follow up to my previous technical questions: I'm having an
argument with a colleague about if it would be better to select fewer
sequences (one per genus or family instead of per species) and build
the tree from that.
I have around 26'000 sequences (~1'000 identical) and a masked
alignment of 1480 sites (nucleotides). (My rule of thumb is to have at
least one variable nucleotide per leaf, though I know this is very
rough.)
Second, if one does a smaller tree, would it then make sense to place
all species in that tree with e.g. EPA-NG to build a complete
reference phylo?
/D
Den ons 30 juni 2021 kl 07:27 skrev Daniel Lundin
<erik.rika...@gmail.com>:
As a follow up to my previous technical questions: I'm having an
argument with a colleague about if it would be better to select fewer
sequences (one per genus or family instead of per species) and build
the tree from that.
I have around 26'000 sequences (~1'000 identical) and a masked
alignment of 1480 sites (nucleotides). (My rule of thumb is to have at
least one variable nucleotide per leaf, though I know this is very
rough.)
Second, if one does a smaller tree, would it then make sense to place
all species in that tree with e.g. EPA-NG to build a complete
reference phylo?
/D
Den ons 30 juni 2021 kl 07:27 skrev Daniel Lundin
<erik.rika...@gmail.com>:

Grimm
Jul 1, 2021, 8:35:59 AM7/1/21
to raxml
Hi Erik,
yes and yes.
For most identification applications we don't need a comprehensive tip set, but a meaningful one. The identical sequences are little to bother because RAxML (at least the classic version, haven't checked for RAxML-ng) eliminates them pre-inference (being useless for tree optimisation).
More annoying are the near-identical, highly similar ones leading to flat terminal subtrees. Probabilistics need something to work with, and a (often random) mutation or two only increase computation time (including bootstrapping). Inference-wise, there's little to gain from a large-sampled flat subtree regarding the deeper relationships (there are good arguments for large samples in other circumstance, e.g. where we want to diminish support for a branching artefact and to better reflect topological ambiguity).
Also, from a theoretical point of view the model assumptions of
probabilistic tree inference (we are optimising mutation probabilities using a dichotomous evolution model, but we look increasingly at fixation probabilities affected by population dynamics) hardly apply the closer we come to the very
leaves of a tree.
So, the much more efficient way is to infer a meaningfully sampled background topology, with the tips being representative for your question, and then place the rest using EPA.
PS EPA will also be negatively affected, in a sense, by too many, too similar tips: you will have placements with low LWR because they are going to be split between the many, near-identical tips; it's not really a big issue because one can easily see the difference between a poorly placed/hard to place query (low LWR but scattered in different subtrees or along the root-proximal tree parts) and one well-placed (equally low LWR but within a clearly distinct subtree)
Here's a screenshot from a table (3 MB large file) how a fully investigated jPlace file can look like (using HTS reads from a plant genus).
My idea was simple: Labelling the main aspects of the backbone tree (Clade x, y subtree, x root, deep branches etc), then use PivotTable to calculate cumulative LWRs (in case the aspect is a subtree not a single tip) and sort that visually to quickly interpret the jPlace result phylogenetically.
Good placing, Guido

Daniel Lundin
Jul 1, 2021, 8:43:07 AM7/1/21
to ra...@googlegroups.com
Thanks for your very informative reply as usual -- Guido,
/D
Den tors 1 juli 2021 kl 14:36 skrev Grimm <grim...@gmail.com>:
>
> Hi Erik,
>
> yes and yes.
>
> For most identification applications we don't need a comprehensive tip set, but a meaningful one. The identical sequences are little to bother because RAxML (at least the classic version, haven't checked for RAxML-ng) eliminates them pre-inference (being useless for tree optimisation).
>
> More annoying are the near-identical, highly similar ones leading to flat terminal subtrees. Probabilistics need something to work with, and a (often random) mutation or two only increase computation time (including bootstrapping). Inference-wise, there's little to gain from a large-sampled flat subtree regarding the deeper relationships (there are good arguments for large samples in other circumstance, e.g. where we want to diminish support for a branching artefact and to better reflect topological ambiguity). Also, from a theoretical point of view the model assumptions of probabilistic tree inference (we are optimising mutation probabilities using a dichotomous evolution model, but we look increasingly at fixation probabilities affected by population dynamics) hardly apply the closer we come to the very leaves of a tree.
>
> So, the much more efficient way is to infer a meaningfully sampled background topology, with the tips being representative for your question, and then place the rest using EPA.
>
> PS EPA will also be negatively affected, in a sense, by too many, too similar tips: you will have placements with low LWR because they are going to be split between the many, near-identical tips; it's not really a big issue because one can easily see the difference between a poorly placed/hard to place query (low LWR but scattered in different subtrees or along the root-proximal tree parts) and one well-placed (equally low LWR but within a clearly distinct subtree)
>
> Here's a screenshot from a table (3 MB large file) how a fully investigated jPlace file can look like (using HTS reads from a plant genus).
> My idea was simple: Labelling the main aspects of the backbone tree (Clade x, y subtree, x root, deep branches etc), then use PivotTable to calculate cumulative LWRs (in case the aspect is a subtree not a single tip) and sort that visually to quickly interpret the jPlace result phylogenetically.
>
> Good placing, Guido
>
>
> To view this discussion on the web visit https://groups.google.com/d/msgid/raxml/855f91cb-68f6-4611-8d4d-46bcfb71c529n%40googlegroups.com.
/D
Den tors 1 juli 2021 kl 14:36 skrev Grimm <grim...@gmail.com>:
>
> Hi Erik,
>
> yes and yes.
>
> For most identification applications we don't need a comprehensive tip set, but a meaningful one. The identical sequences are little to bother because RAxML (at least the classic version, haven't checked for RAxML-ng) eliminates them pre-inference (being useless for tree optimisation).
>
> More annoying are the near-identical, highly similar ones leading to flat terminal subtrees. Probabilistics need something to work with, and a (often random) mutation or two only increase computation time (including bootstrapping). Inference-wise, there's little to gain from a large-sampled flat subtree regarding the deeper relationships (there are good arguments for large samples in other circumstance, e.g. where we want to diminish support for a branching artefact and to better reflect topological ambiguity). Also, from a theoretical point of view the model assumptions of probabilistic tree inference (we are optimising mutation probabilities using a dichotomous evolution model, but we look increasingly at fixation probabilities affected by population dynamics) hardly apply the closer we come to the very leaves of a tree.
>
> So, the much more efficient way is to infer a meaningfully sampled background topology, with the tips being representative for your question, and then place the rest using EPA.
>
> PS EPA will also be negatively affected, in a sense, by too many, too similar tips: you will have placements with low LWR because they are going to be split between the many, near-identical tips; it's not really a big issue because one can easily see the difference between a poorly placed/hard to place query (low LWR but scattered in different subtrees or along the root-proximal tree parts) and one well-placed (equally low LWR but within a clearly distinct subtree)
>
> Here's a screenshot from a table (3 MB large file) how a fully investigated jPlace file can look like (using HTS reads from a plant genus).
> My idea was simple: Labelling the main aspects of the backbone tree (Clade x, y subtree, x root, deep branches etc), then use PivotTable to calculate cumulative LWRs (in case the aspect is a subtree not a single tip) and sort that visually to quickly interpret the jPlace result phylogenetically.
>
> Good placing, Guido
>
>
Grimm
Jul 1, 2021, 10:32:45 AM7/1/21
to raxml
Hej Daniel,
Ingen ursak :) It's a kind of hobby of mine to help out with practical problems like this.
A note regarding the selection of the representative tip set: we always fared lighter with random placeholder selection during review, I suppose because it strikes one more objective. In case of your particular setting (26,000 tips x 1500 basepairs), if aiming for genus-level assignments I would however go for at least including the two most divergent tips e.g. per generic lineage (or target taxon). This way, if EPA places the query next to either tip representing the genus, it points to being an acutal member of the genus (taxon), while placed at the genus' root it could also be a non-sampled sibling genus.
It can also be interesting to filter the problematic/or deeper-than-genus-root EPA placed queries and use them to run another focussed tree to see where they place if included in the analysis without having to re-infer the whole 26k-tip tree and waste computation time on its trivial
(genetically-phylogenetically obvious)
aspects
We used this logic recently to assess the phylogenetic placement of our somewhat pseudogenic 5S-IGS lineage in beeches; low-abundant variants EPA placed in the nether-regions of our reference tree:
[Note: It's still only a pre-print because we need to make it more "generally appealing" before resubmitting. But the Results part will remain largely unchanged since it and the methodological set-up involving EPA to identify sequence affinity fared well with the reviewers.]
Another nice thing about using EPA for idendification instead of inferring a 28k-tip tree is that one can also quickly filter subsets, e.g. accessions probably belonging to a certain family to then run (much quicker) intra-family trees. With the SuperTree (or SuperNetwork, but they more easily hit inference limitations) approaches, one could even combine the backbone and any family-level (outgroup-free) or other aspect-wise tree into an all-inclusive tree (if required). They are also much quicker to bootstrap in case one has topological ambiguity issues. So, there's really a lot one can do with the saved inference time to focus on puzzling aspects if needed.
/G
Alexandros Stamatakis
Jul 1, 2021, 11:01:13 PM7/1/21
to ra...@googlegroups.com
Thank you very much Guido :-)
Nothing to add from my side.
Alexis
On 01.07.21 17:32, Grimm wrote:
> Hej Daniel,
>
> Ingen ursak :) It's a kind of hobby of mine to help out with practical
> problems like this.
>
> A note regarding the selection of the representative tip set: we always
> fared lighter with random placeholder selection during review, I suppose
> because it strikes one more objective. In case of your particular
> setting (26,000 tips x 1500 basepairs), if aiming for genus-level
> assignments I would however go for at least including the two most
> divergent tips e.g. per generic lineage (or target taxon). This way, if
> EPA places the query next to either tip representing the genus, it
> points to being an acutal member of the genus (taxon), while placed at
> the genus' root it could also be a non-sampled sibling genus.
>
> It can also be interesting to filter the problematic/or
> deeper-than-genus-root EPA placed queries and use them to run another
> focussed tree to see where they place if included in the analysis
> without having to re-infer the whole 26k-tip tree and waste computation
> time on its trivial (genetically-phylogenetically obvious) aspects
>
> We used this logic recently to assess the phylogenetic placement of our
> somewhat pseudogenic 5S-IGS lineage in beeches; low-abundant variants
> EPA placed in the nether-regions of our reference tree:
>
> Cardoni et al., 2021, bioRxiv <http://dx.doi.org/10.1101/2021.02.26.433057>
> >> > > www.exelixis-lab.org <http://www.exelixis-lab.org>
> <https://groups.google.com/d/msgid/raxml/855f91cb-68f6-4611-8d4d-46bcfb71c529n%40googlegroups.com>.
> <https://groups.google.com/d/msgid/raxml/8c5612ae-0814-444c-a941-4e4ec6eb19a3n%40googlegroups.com?utm_medium=email&utm_source=footer>.
Nothing to add from my side.
Alexis
On 01.07.21 17:32, Grimm wrote:
> Hej Daniel,
>
> Ingen ursak :) It's a kind of hobby of mine to help out with practical
> problems like this.
>
> A note regarding the selection of the representative tip set: we always
> fared lighter with random placeholder selection during review, I suppose
> because it strikes one more objective. In case of your particular
> setting (26,000 tips x 1500 basepairs), if aiming for genus-level
> assignments I would however go for at least including the two most
> divergent tips e.g. per generic lineage (or target taxon). This way, if
> EPA places the query next to either tip representing the genus, it
> points to being an acutal member of the genus (taxon), while placed at
> the genus' root it could also be a non-sampled sibling genus.
>
> It can also be interesting to filter the problematic/or
> deeper-than-genus-root EPA placed queries and use them to run another
> focussed tree to see where they place if included in the analysis
> without having to re-infer the whole 26k-tip tree and waste computation
> time on its trivial (genetically-phylogenetically obvious) aspects
>
> We used this logic recently to assess the phylogenetic placement of our
> somewhat pseudogenic 5S-IGS lineage in beeches; low-abundant variants
> EPA placed in the nether-regions of our reference tree:
>
> >> > >
> >> > > --
> >> > > You received this message because you are subscribed to the
> Google Groups "raxml" group.
> >> > > To unsubscribe from this group and stop receiving emails
> from it, send an email to raxml+un...@googlegroups.com.
> >> > > To view this discussion on the web visit
> https://groups.google.com/d/msgid/raxml/54b2e209-bb5a-035e-e29c-bb5f5e8bdad4%40gmail.com
> <https://groups.google.com/d/msgid/raxml/54b2e209-bb5a-035e-e29c-bb5f5e8bdad4%40gmail.com>.
> >> > > --
> >> > > You received this message because you are subscribed to the
> Google Groups "raxml" group.
> >> > > To unsubscribe from this group and stop receiving emails
> from it, send an email to raxml+un...@googlegroups.com.
> >> > > To view this discussion on the web visit
> https://groups.google.com/d/msgid/raxml/54b2e209-bb5a-035e-e29c-bb5f5e8bdad4%40gmail.com
>
> >
> > --
> > You received this message because you are subscribed to the
> Google Groups "raxml" group.
> > To unsubscribe from this group and stop receiving emails from it,
> send an email to raxml+un...@googlegroups.com.
> > To view this discussion on the web visit
> https://groups.google.com/d/msgid/raxml/855f91cb-68f6-4611-8d4d-46bcfb71c529n%40googlegroups.com
> >
> > --
> > You received this message because you are subscribed to the
> Google Groups "raxml" group.
> > To unsubscribe from this group and stop receiving emails from it,
> send an email to raxml+un...@googlegroups.com.
> > To view this discussion on the web visit
> <https://groups.google.com/d/msgid/raxml/855f91cb-68f6-4611-8d4d-46bcfb71c529n%40googlegroups.com>.
>
>
> --
> You received this message because you are subscribed to the Google
> Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to raxml+un...@googlegroups.com
> <mailto:raxml+un...@googlegroups.com>.
>
> --
> You received this message because you are subscribed to the Google
> Groups "raxml" group.
> To unsubscribe from this group and stop receiving emails from it, send
> an email to raxml+un...@googlegroups.com
> To view this discussion on the web visit
> https://groups.google.com/d/msgid/raxml/8c5612ae-0814-444c-a941-4e4ec6eb19a3n%40googlegroups.com
> <https://groups.google.com/d/msgid/raxml/8c5612ae-0814-444c-a941-4e4ec6eb19a3n%40googlegroups.com?utm_medium=email&utm_source=footer>.
Daniel Lundin
Jul 3, 2021, 1:21:49 AM7/3/21
to ra...@googlegroups.com
Den tors 1 juli 2021 kl 16:32 skrev Grimm <grim...@gmail.com>:
>
> Hej Daniel,
>
> Ingen ursak :) It's a kind of hobby of mine to help out with practical problems like this.
>
> A note regarding the selection of the representative tip set: we always fared lighter with random placeholder selection during review, I suppose because it strikes one more objective. In case of your particular setting (26,000 tips x 1500 basepairs), if aiming for genus-level assignments I would however go for at least including the two most divergent tips e.g. per generic lineage (or target taxon). This way, if EPA places the query next to either tip representing the genus, it points to being an acutal member of the genus (taxon), while placed at the genus' root it could also be a non-sampled sibling genus.
>
OK. I clustered the sequences at 85% identity to get a set of slightly
>
> Hej Daniel,
>
> Ingen ursak :) It's a kind of hobby of mine to help out with practical problems like this.
>
> A note regarding the selection of the representative tip set: we always fared lighter with random placeholder selection during review, I suppose because it strikes one more objective. In case of your particular setting (26,000 tips x 1500 basepairs), if aiming for genus-level assignments I would however go for at least including the two most divergent tips e.g. per generic lineage (or target taxon). This way, if EPA places the query next to either tip representing the genus, it points to being an acutal member of the genus (taxon), while placed at the genus' root it could also be a non-sampled sibling genus.
>
more than 1000 sequences. I do this often when working with protein
sequences to get a set that is representative of "sequence space".
> It can also be interesting to filter the problematic/or deeper-than-genus-root EPA placed queries and use them to run another focussed tree to see where they place if included in the analysis without having to re-infer the whole 26k-tip tree and waste computation time on its trivial (genetically-phylogenetically obvious) aspects
>
> We used this logic recently to assess the phylogenetic placement of our somewhat pseudogenic 5S-IGS lineage in beeches; low-abundant variants EPA placed in the nether-regions of our reference tree:
>
> Cardoni et al., 2021, bioRxiv
>
> [Note: It's still only a pre-print because we need to make it more "generally appealing" before resubmitting. But the Results part will remain largely unchanged since it and the methodological set-up involving EPA to identify sequence affinity fared well with the reviewers.]
>
> Another nice thing about using EPA for idendification instead of inferring a 28k-tip tree is that one can also quickly filter subsets, e.g. accessions probably belonging to a certain family to then run (much quicker) intra-family trees. With the SuperTree (or SuperNetwork, but they more easily hit inference limitations) approaches, one could even combine the backbone and any family-level (outgroup-free) or other aspect-wise tree into an all-inclusive tree (if required). They are also much quicker to bootstrap in case one has topological ambiguity issues. So, there's really a lot one can do with the saved inference time to focus on puzzling aspects if needed.
>
> /G
>
Grimm
Jul 5, 2021, 2:42:31 AM7/5/21
to raxml
Re: "I clustered the sequences at 85% identity to get a set of slightly more than 1000 sequences. I do this often when working with protein
sequences to get a set that is representative of "sequence space"." – in case you want to try non-fixed (but phylogeny-aware) thresholds in defining e.g. genus-level groups, Markus Göker's OPTSIL (http://www.goeker.org/mg/clustering/) may be worth a look, too.
The program OPTSIL is a [platform-independent, freeware] tool for the optimization of threshold-based linkage
clustering runs. It was developed for molecular taxonomy, even though other
applications are possible. The typical usage is to conduct clusterings for a
series of distance thresholds, and to calculate the agreement between each
resulting partition and one or several reference partitions. The optimal
threshold values are those for which the agreement is highest. The whole range
of linkage clustering approaches from single linkage to complete linkage can be
optimized.
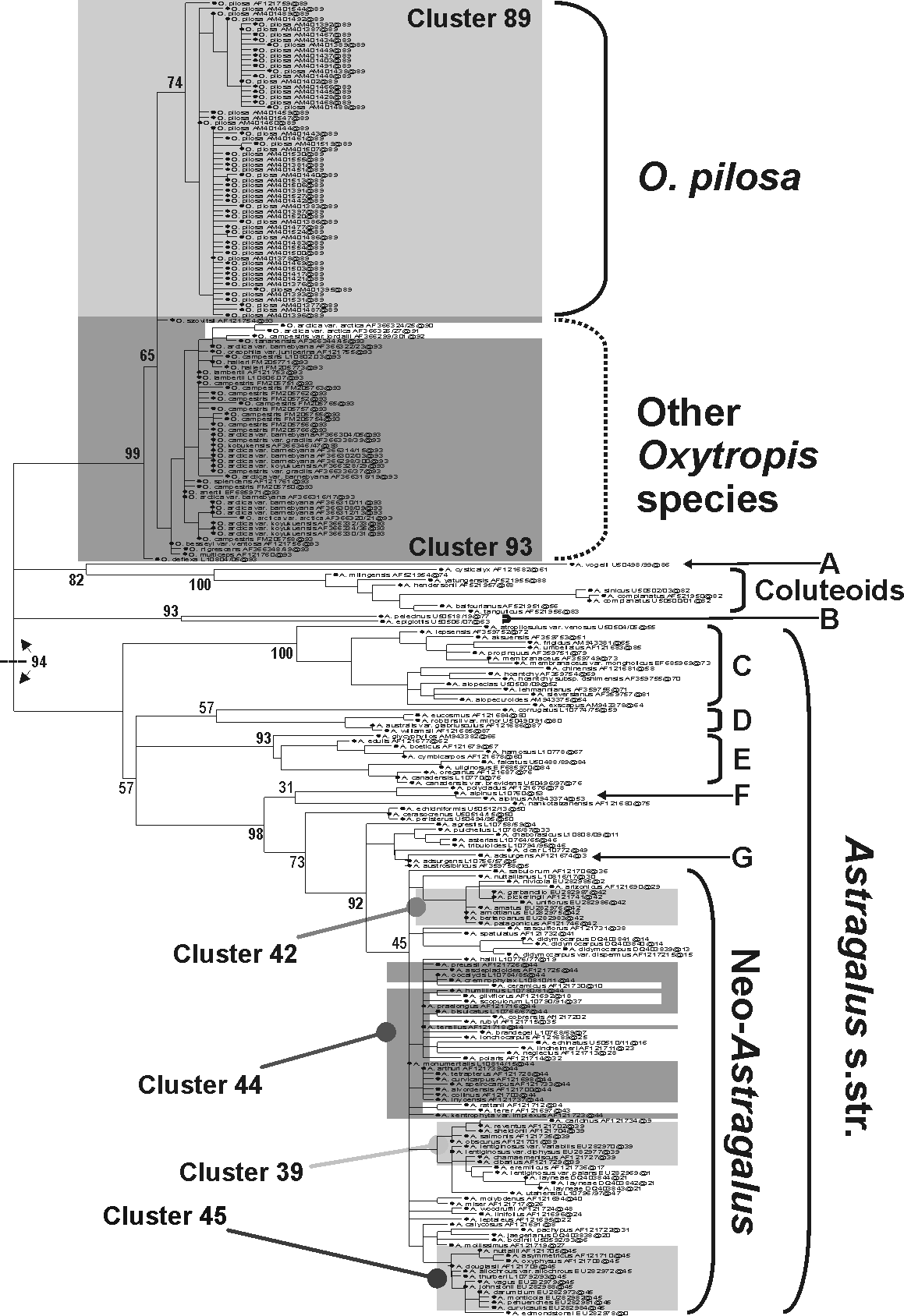
Identifying reference groups for placing large sequence samples with EPA would be one of those "other applications possible", e.g. to judge how many genus-level taxa you have in the 1000-sequence subset to put equal-value labels on each of the 1000 reference tips. There are several key (early) papers linked on the page showing different application angles (including one of those, where Markus and I teamed up to solve a fellow's problem: Schlee et al. 2010, main figure below, where we "objectively compare[d] biodiversity (given the limits of the
respective molecular marker used)") Funding constraints prevented we could do more down that alley.
/G
Reply all
Reply to author
Forward
0 new messages