Workflow for counting cells with multiple fluorophores and puncta

achamess
Hi,
QuPath looks like it could be an extremely useful tool for a standard analysis I do.
Basically, I want to:
- Identify cells in spinal cord tissue sections, either automatically or by manually.
- I'm willing to draw ROIs by hand or some combination of auto-detect plus clean-up by hand. That's not horrible. But counting and finding overlaps, that is the part that I need to automate.
- Identify the cellular staining: Usually, cells have (DAPI) and then one or more IHC signals (antibody) or ISH signal (RNAscope, which usually show as puncta)
- Count the overlapping events: I want to see which cells have combinations of fluorescent signals: red alone, red+green, green alone, etc. Sometimes this will be binary (yes/no) but it'd be nice to make more bins like absent, low, medium and high.
- Optional for ISH: Count puncta inside of cells.
This is such a core analysis for a lot of neuroscience. I've tried to use Fiji and I still don't know how to do this in a straight forward way. I've resorted to just using Photoshop and manually counting, but that's a huge pain.
CellProfiler looks like it might have some cool features, and I presented the problem there too:
http://forum.cellprofiler.org/t/rnascope-in-spinal-cord-neurons-quantitation-of-overlap-between-different-in-situ-probes/5355/3
But could some combination of QuPath + Cell Profiler (or QuPath alone) get the job done? I feel like it can.
Here is an example image:

Any help would be greatly appreciated.
Note: I cross posted this on Github and @svidro gave a great response. But I'm putting it here to get more people involved.

micros...@gmail.com
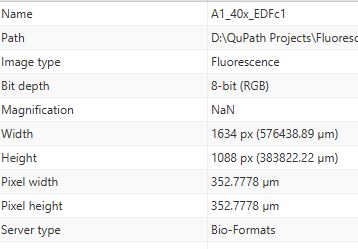
The starting .nd2 file runs fine, but the adjusted files... well I am having trouble getting 4 channels into a TIFF that QuPath can read.
I will give Fiji a try, but I don't have much experience editing metadata. Anyone else have any ideas?

Pete
- Is there already a CellProfiler solution? If so, https://github.com/qupath/qupath/issues/123 might be interesting. But in that case, would introducing QuPath help - or have you already got the result you need?
- Something looks badly wrong in the metadata of microscopyra's screenshot (not just the RGB-ness, but also the pixel size), however I don't know where the .nd2 file came from. Was it posted somewhere?
- Check out http://forum.imagej.net/t/counting-double-labeled-cells-in-fiji/3832/2 - that would be my suggestion if you'd like to try it in QuPath
achamess
- Is there already a CellProfiler solution? If so, https://github.com/qupath/qupath/issues/123 might be interesting. But in that case, would introducing QuPath help - or have you already got the result you need?
- Something looks badly wrong in the metadata of microscopyra's screenshot (not just the RGB-ness, but also the pixel size), however I don't know where the .nd2 file came from. Was it posted somewhere?
- Check out http://forum.imagej.net/t/counting-double-labeled-cells-in-fiji/3832/2 - that would be my suggestion if you'd like to try it in QuPath
micros...@gmail.com
"Unfortunately, it will take quite a bit of work to set the images up in ImageJ, as you have to import all 4 base images, split them all (Image-Stacks-Stack to images), edit the pixel size (Image-Properties), close all of the blank channels, then remerge them (Image-Stack-Images to stack), and finally create a composite (Image-Color-Merge channels). You may end up wanting to ask on the ImageJ forums about writing a macro script for that, as it will require quite a bit of picking out the right file names, and which channels to keep based on the file names... Not sure how many images you want to do this for, but at least this should work."
I think he ended up going with the original .nd2 file which only had data from 3 channels, but apparently that was enough. It also turned out that I originally couldn't open the OME-TIFF (or the .nd2) generated by the above method until I updated my version of the bioformats jar, which hadn't been done in about 6 months or more. Keeping up with those updates is important!
achamess
micros...@gmail.com
- If I make automatic detections, which of the many measurements is the one that tells me how much signal is inside my cell? The sum or mean or what? What's the best way to see how much signal intensity is inside a detection?
- Is there a way to turn hand drawn annotations into detections? Alternatively, if I use the automated segmentation on DAPI, can I go and amend some of the detections manually?
This turns the annotation into a detection object, but NOT a cell. I don't know of any way to draw a nucleus+cytoplasm, and I do not think the program is designed to allow editing of the detections. As Pete just reminded me, annotations have their associated values updated dynamically, while detections have fixed measurements when they are created. For example, if you did manage to edit a cell boundary, you would not be changing the measurement for "Cell: Area."
- Is there a way to identify whole cell boundaries, not just nuclei?
achamess
The sum should be the total intensity in each pixel, and is great for Nuclear only measurements. Unfortunately it is not included for the Cytoplasm when the cells are generated. "Cell: Channel X mean" (or Cytoplasm) is probably your best bet if you do not want to generate other measurements. You could also multiple the "Cell: Channel X mean" by the "Cell: Area" to get the whole cell sum, and then subtract the "Nucleus: Channel X sum" to get the cytoplasm sum if needed. I tend to think best is related to the specific experiment, but maybe Pete will have some better ideas!
Sort of, see the final post of: https://groups.google.com/forum/#!topic/qupath-users/JBrVa0DQ8pk
This turns the annotation into a detection object, but NOT a cell. I don't know of any way to draw a nucleus+cytoplasm, and I do not think the program is designed to allow editing of the detections. As Pete just reminded me, annotations have their associated values updated dynamically, while detections have fixed measurements when they are created. For example, if you did manage to edit a cell boundary, you would not be changing the measurement for "Cell: Area."


If you mean trying to use image information to create the cell boundaries, there is one experimental method (for H-DAB only I think?) called Cell+membrane detection, but I had mixed results with that. Otherwise, I think it is up to the "Cell expansion" parameter when you are performing the segmentation. One of the reasons I like the subcellular detection command for IHC is that you can increase the size of the cells dramatically without the extra "empty" space influencing the measurement like it would with a cell mean or cytoplasm mean intensity. Subcellular detection isn't just for spot counting, it can be used as more of a general "threshold within a cell" measurement.

micros...@gmail.com
So ultimately I see QuPath getting me part of the way with automatic detection and then I will go in and fix the obviously wrong ones. But it doesn't seem like that's really an option. In other scenarios, the nucleus and the cell are very different, and I see now way to do this but by hand (draw borders/ROIs around cells).
But you've also mentioned that the empty space (areas inside ROI/detection without channel signal) influence that measure. What if we background subtract? Is there a way to do that? Or would dividing the sum by the cross sectional area (a derived metric) be better? Also, what is the best way to set a threshold in QuPath? Using the brightness/contrast?
So for this second kind of image, with these big round cells that I plan to draw by hand, is there a way for me to get the metrics I want? Forget the automated segmentation. Basically I just want to draw cells and then count puncta/spots inside. It seems like the subcellular detection is the way to go, but I don't know how to get this to work with the annotations. I open up that dialog box and nothing happens.
achamess
Not sure how much you have played around with the settings for the nuclear detection, but it looks like in the spinal cord image your threshold might be a little bit low. I just played around with the other .nd2 file, and a decent threshold was around 1200-1500. I assume, though, you would want to aim low since while it won't work very well to draw in new cells by hand, it's quite easy to manually delete cells, or select a subset based on some criteria (size, shape, intensity) and delete them.
Any type of measurement will be subject to both background and space, which is why it is so dependent on the experiment. If you are looking at a single spot in a very large cross section of a nuclei, vs a single spot in a very narrow tip of a nuclei, your mean is going to be very different. On the other hand, if your background is strong, an empty "large" nuclei might sum up to the same intensity as a single spot.
Subcellular detections can only be performed on cells, and they require pixel width and height values in order to be run. With either of those things missing, it will not run. In your case, you don't have cells, you have annotations. You can use the script previously linked to turn them into detections, but I am not sure how to fool the program into thinking they are cell objects, or what problems it might cause if you were able to (cell objects assume nucleus and cytoplasm, which you can see if you reduce the "Cell expansion" to 0 in cell detection. You no longer get cells, just detection objects).
For the last bit, I think the easiest method would be to use the ImageJ macro runner due to wanting to detect multiple channels at once. Your images are fairly small, so you would not need to downsample much, if at all, to drop them into ImageJ, remove the green channel maybe, and then perform a segmentation on a grayscale version of the no-green image (Analyze->Analyze Particles, along with Plugins->Send overlay to QuPath-Detections). Once those detection measurements are back in QuPath, you could generate measurements like "Blue mean" "Red Mean" etc. and classify off of that. At least based on what I think you are wanting. You could also segment WITH green, but that would be far messier, and you would probably want to remove some of the segmentation after getting it back into QuPath based on shape, size or color.
If you wanted to hand draw it, you could still do that, convert the annotations into detections, and then generate all of the measurements you need through Analyze-> Calculate Features-> Add intensity features. For hundreds of images, the ImageJ Extension would be very useful, but for just a few it would probably work well enough. Either way, once you were done, you would probably want to draw an annotation region around the whole thing so you could use Show Annotation Measurements to summarize.
On the other hand... I can't tell from the image but if the blue is just DAPI, and all of the cells have detectable nuclei, you could just do the nuclear segmentation with a 0.5-1um Cell expansion in order to make sure you only pick up the signal from that cell. Then the "Cytoplasm: Channel X mean intensity" should work just fine.
micros...@gmail.com
What's a good way to systematically find a reasonable range for thresholds?
If you want a less biased measure of background, you could hand draw an annotation area somewhere within the sample but without any nuclei and use Calculate Intensity Features on that (generate the mean/min/max/std dev).
Background substracting will need to happen outside of QuPath, right?
When I use the annotation -> detection script, I can then use the 'calculate intensity' function. But what about sum? Can I get that?
For the last bit, I think the easiest method would be to use the ImageJ macro runner due to wanting to detect multiple channels at once. Your images are fairly small, so you would not need to downsample much, if at all, to drop them into ImageJ, remove the green channel maybe, and then perform a segmentation on a grayscale version of the no-green image (Analyze->Analyze Particles, along with Plugins->Send overlay to QuPath-Detections). Once those detection measurements are back in QuPath, you could generate measurements like "Blue mean" "Red Mean" etc. and classify off of that. At least based on what I think you are wanting. You could also segment WITH green, but that would be far messier, and you would probably want to remove some of the segmentation after getting it back into QuPath based on shape, size or color.Could you clarify. What is this for? For detecting cells?
Can I get the sum somehow? Or am I misunderstanding that? I agree that the subcellular detection is the best way to figure out how much signal is into a cell, since it doesn't count the blank space. For IHC, where the cell is full, this isn't as much as an issue, but even then, I think you've suggested that the subcellular detection could still be used to count the whole internal contents of the cell as one big spot. Yes?
And no problem, this is all for fun :)
Pete
Pete: Is there any way to turn a detection into a cell object?
achamess
import qupath.lib.objects.PathTileObjectimport qupath.lib.roi.RectangleROIimport qupath.lib.scripting.QPimport qupath.lib.objects.PathCellObject
// Set this to true to use the bounding box of the ROI, rather than the ROI itselfboolean useBoundingBox = false
// Get the current hierarchydef hierarchy = QP.getCurrentHierarchy()
// Get the select objectsdef selected = getAnnotationObjects()
// Check we have anything to work withif (selected.isEmpty()) { print("No objects selected!") return}
// Loop through objectsdef newDetections = new ArrayList<>()for (def pathObject in selected) {
// Unlikely to happen... but skip any objects not having a ROI if (!pathObject.hasROI()) { print("Skipping object without ROI: " + pathObject) continue }
// Don't create a second annotation, unless we want a bounding box if (!useBoundingBox && pathObject.isDetection()) { print("Skipping annotation: " + pathObject) continue }
// Create an annotation for whichever object is selected, with the same class // Note: because ROIs are (or should be) immutable, the same ROI is used here, rather than a duplicate def roi = pathObject.getROI() if (useBoundingBox) roi = new RectangleROI( roi.getBoundsX(), roi.getBoundsY(), roi.getBoundsWidth(), roi.getBoundsHeight(), roi.getC(), roi.getZ(), roi.getT()) def detection = new PathCellObject(roi,null,pathObject.getPathClass()) newDetections.add(detection) print("Adding " + detection)}
// Actually add the objectshierarchy.addPathObjects(newDetections, false)if (newDetections.size() > 1) print("Added " + newDetections.size() + " detections(s)")And here is the output:
INFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonINFO: Adding PolygonERROR: Error at line 58: null
ERROR: Script error at qupath.lib.objects.hierarchy.PathObjectHierarchy.addPathObjectToList(PathObjectHierarchy.java:350) at qupath.lib.objects.hierarchy.PathObjectHierarchy.addPathObjects(PathObjectHierarchy.java:519) at qupath.lib.objects.hierarchy.PathObjectHierarchy$addPathObjects.call(Unknown Source) at org.codehaus.groovy.runtime.callsite.CallSiteArray.defaultCall(CallSiteArray.java:48) at org.codehaus.groovy.runtime.callsite.AbstractCallSite.call(AbstractCallSite.java:113) at org.codehaus.groovy.runtime.callsite.AbstractCallSite.call(AbstractCallSite.java:133) at Script10.run(Script10.groovy:59) at org.codehaus.groovy.jsr223.GroovyScriptEngineImpl.eval(GroovyScriptEngineImpl.java:343) at org.codehaus.groovy.jsr223.GroovyScriptEngineImpl.eval(GroovyScriptEngineImpl.java:152) at qupath.lib.scripting.DefaultScriptEditor.executeScript(DefaultScriptEditor.java:765) at qupath.lib.scripting.DefaultScriptEditor.executeScript(DefaultScriptEditor.java:695) at qupath.lib.scripting.DefaultScriptEditor.executeScript(DefaultScriptEditor.java:677) at qupath.lib.scripting.DefaultScriptEditor.access$400(DefaultScriptEditor.java:136) at qupath.lib.scripting.DefaultScriptEditor$2.run(DefaultScriptEditor.java:1029) at java.util.concurrent.Executors$RunnableAdapter.call(Executors.java:511) at java.util.concurrent.FutureTask.run(FutureTask.java:266) at java.util.concurrent.ThreadPoolExecutor.runWorker(ThreadPoolExecutor.java:1142) at java.util.concurrent.ThreadPoolExecutor$Worker.run(ThreadPoolExecutor.java:617) at java.lang.Thread.run(Thread.java:745)
Pete
micros...@gmail.com
I went ahead and wrote a test case that works be picking all annotations that are not on the "outside/top" layer so that you can have your tissue annotation drawn (possibly in multiple parts) and then convert all annotations within the largest annotations into detections. If you don't want to have an outer tissue annotation, the previous script will work fine, though. This might be more useful for anyone areas out of brightfield tissue samples and running subcellular detection on them. Playing around with it a little, the speed of running subcellular detection blows away the positive pixel detection, does not run into threshold or divide by 0 problems, and does not seem to run into problems with needing tiling. It does require slightly more calculations to get the percent positive, but that's fairly trivial at this point.
import qupath.lib.objects.PathCellObject
// Get the current hierarchy
def hierarchy = getCurrentHierarchy()
// Get the select objects
def targets = getObjects{return it.getLevel()!=1 && it.isAnnotation()}
// Check we have anything to work with
if ( targets.isEmpty()) {
print("No objects selected!")
return
}
// Loop through objects
def newDetections = new ArrayList<>()
for (def pathObject in targets) {
// Unlikely to happen... but skip any objects not having a ROI
if (!pathObject.hasROI()) {
print("Skipping object without ROI: " + pathObject)
continue
}
def roi = pathObject.getROI()
def detection = new PathCellObject(roi,roi,pathObject.getPathClass())
newDetections.add(detection)
print("Adding " + detection)
}
removeObjects( targets, true)
// Actually add the objects
hierarchy.addPathObjects(newDetections, false)
//Remove nucleus ROI
newDetections.each{it.nucleus = null}
fireHierarchyUpdate()
if (newDetections.size() > 1)
print("Added " + newDetections.size() + " detections(s)")
Positive pixel detection on results, showing eosin subcellular detections in a large region.

Pete
Looks good, I get functional large pathCellObjects that I can run Subcellular spot detection in, which is amazing. Is there a reason to eliminate the nucleus ROI? I do not see it show up in the hierarchy at least, though I suppose it does create duplicate measurements when adding area to the cell. Either way seems to work with subcellular detections.

abou...@stanford.edu
micros...@gmail.com

fuhy...@gmail.com

micros...@gmail.com
micros...@gmail.com
abou...@stanford.edu
micros...@gmail.com
//Adjust downsample if ImageJ macro runner generates area size errors.
double downsample = 1
createSelectAllObject(true);
import qupath.imagej.plugins.ImageJMacroRunner
import qupath.lib.plugins.parameters.ParameterList
// Create a macro runner so we can check what the parameter list contains
def params = new ImageJMacroRunner(getQuPath()).getParameterList()
print ParameterList.getParameterListJSON(params, ' ')
// Change the value of a parameter, using the JSON to identify the key
params.getParameters().get('downsampleFactor').setValue(downsample)
print ParameterList.getParameterListJSON(params, ' ')
// Get the macro text and other required variables
def macro = 'run("8-bit");run("Gaussian Blur...", "sigma=4");setOption("BlackBackground", true);setAutoThreshold("Default");run("Threshold...");setThreshold(1, 255);run("Convert to Mask");run("Fill Holes");run("Close");run("Dilate");run("Dilate");run("Dilate");run("Dilate");run("Dilate");run("Dilate");run("Erode");run("Erode");run("Erode");run("Erode");run("Erode");run("Erode");run("Analyze Particles...", "size=1000-Infinity add");roiManager("Select", 0);run("Send ROI to QuPath");close();'
def imageData = getCurrentImageData()
def annotations = getAnnotationObjects()
// Loop through the annotations and run the macro
for (annotation in annotations) {
ImageJMacroRunner.runMacro(params, imageData, null, annotation, macro)
}
print 'Done!'
annotations = getAnnotationObjects();
removeObject(annotations[0], true)