
AnnaC
Hello everyone,
I am trying to understand chimera checking. I have several doubts and I would really appreciate if someone can help me to understand a little bit better these facts.
1) Which algorithm works better for length around 270-320bp? We are using Ion Torrent so we cannot arrive at 400bp minimum length…
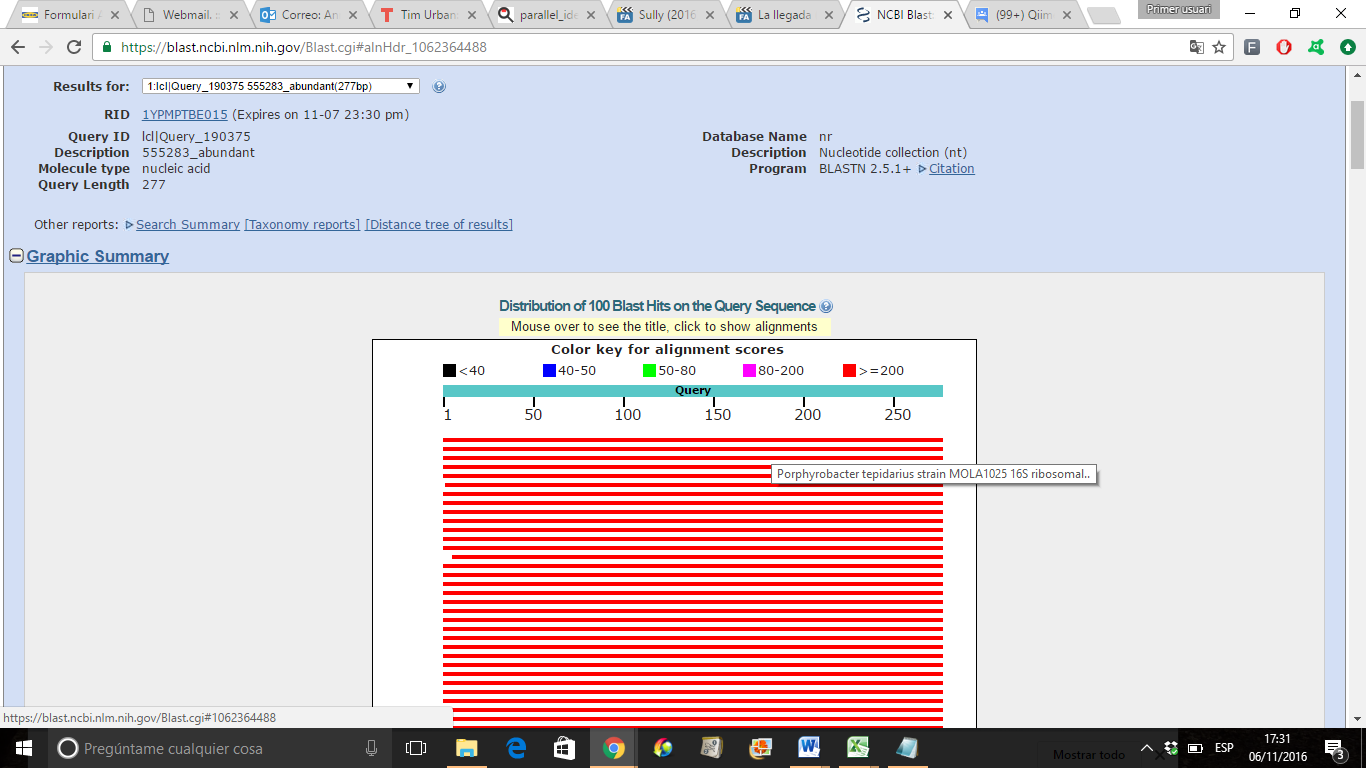
(2) 2) I have been doing some test and I feel surprised and confused when a sequence like that is considered a chimera using ChimeraSlayer (blast screenshot attached). BLAST results: https://blast.ncbi.nlm.nih.gov/Blast.cgi#alnHdr_1083262127
(3) 3) One specific chimera OTU would represent really low abundant pattern, wouldn’t be? If it is like this, it is not better to remove only really low abundant OTUs rather than remove putative chimera that they do not look like chimeras?
(4) 4) I am working on several skin sites and I have seen some “putative chimeras” that were exclusive to some individual and in different skin sites… So I wouldn’t think they are actual chimeras, but some strain or species.
Could anyone help me to better understand these points?
Thank you so much!!!
Anna


{kind=link}