PIGx ChIP-seq pipeline error

Elizavet...@mdc-berlin.de
Dear PIGx ChIP-seq pipeline developers,
Here is the error message for the PIGx-ChIP-seq:

How I can fix this error?
Best,
MDC, Bunina lab

Altuna Akalin
--
You received this message because you are subscribed to the Google Groups "pigx" group.
To unsubscribe from this group and stop receiving emails from it, send an email to pigx+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/pigx/042894414eff4bb2839d2e769f846c3e%40mdc-berlin.de.

Bora Uyar
--
You received this message because you are subscribed to the Google Groups "pigx" group.
To unsubscribe from this group and stop receiving emails from it, send an email to pigx+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/pigx/042894414eff4bb2839d2e769f846c3e%40mdc-berlin.de.
_____________
Dr. Bora Uyar
Bioinformatics Scientist
Bioinformatics and Omics Data Science
Max Delbrueck Center (MDC) for Molecular Medicine
The Berlin Institute for Medical Systems Biology (BIMSB):
Hannoversche Str. 28, 10115 Berlin
email: bora...@mdc-berlin.de
mobile: +49 172 949 5680
Elizavet...@mdc-berlin.de
Thank you very much, I apologise for such a silly mistake. Another (probably) silly question: how do I fix these mistakes? Peaks1 and peaks2 are calculations from two separate studies, each containing several control samples and several input samples.

Отправлено: 12 октября 2022 г. 14:05:59
Кому: Kulaeva, Elizaveta
Копия: pi...@googlegroups.com
Тема: [ext] Re: PIGx ChIP-seq pipeline error
Elizavet...@mdc-berlin.de
And also how I can delete whitespaces from the fasta genome file?
Отправлено: 12 октября 2022 г. 14:17:31
Кому: Bora Uyar
Копия: pi...@googlegroups.com
Тема: Re: [ext] Re: PIGx ChIP-seq pipeline error
Bora Uyar
Elizavet...@mdc-berlin.de
Best,
Lisa
Отправлено: 12 октября 2022 г. 14:22:59
Bora Uyar
Bora Uyar
Elizavet...@mdc-berlin.de
Now ( because of modification to the fasta headers) there are problems with the gtf file:

What should I do now?
Lisa
Отправлено: 12 октября 2022 г. 14:46:01
Bora Uyar

Alexander Blume
You also need to modify the gtf annotation file using:
sed '/^#/d' annotation_file.gtf > annotation_file_no_header.gtf
Best,
Alex
> On 12. Oct 2022, at 15:07, Bora Uyar <borauy...@gmail.com> wrote:
>
> You would need to check how your fasta headers look and how the chromosomes are represented in the GTF file.
> First you would need to figure out what is the mismatch (is it the naming convention? e.g. chrI vs I) or is it that the GTF file contains more chromosomes than available in the fasta file?
>
>
> On Wed, Oct 12, 2022 at 2:56 PM Elizavet...@mdc-berlin.de <Elizavet...@mdc-berlin.de> wrote:
> Thank you for all your help, and I apologise for asking so many questions :(
>
> Now ( because of modification to the fasta headers) there are problems with the gtf file:
> What should I do now?
>
> Lisa
>
>
> Отправлено: 12 октября 2022 г. 14:46:01
> Кому: Kulaeva, Elizaveta
> Копия: pi...@googlegroups.com
> Тема: Re: [ext] Re: PIGx ChIP-seq pipeline error
>
> and you can save it into a file like this
> ```
> cat test.fasta | sed -E "s|(^>.+) .+$|\\1|g" > test.out.fasta
> ```
>
> On Wed, Oct 12, 2022 at 2:45 PM Bora Uyar <borauy...@gmail.com> wrote:
> I assume you have a fasta file (test.fasta) that looks like this
> ```
> >chr1 SomeAnnotation
> ATGATA
>
> >chr2 Sometext
> AGATACAT
> ```
>
> Basically, as far as I know, the pipeline wants you to have a fasta file with just the chromosome names.
> You can do that like this
>
> ```
> cat test.fasta | sed -E "s|(^>.+) .+$|\\1|g"
>
> ```
> The command will strip off anything after the first space encountered on fasta headers.
> Best,
> Bora
>
>
>
>
>
>
>
>
>
> On Wed, Oct 12, 2022 at 2:34 PM Elizavet...@mdc-berlin.de <Elizavet...@mdc-berlin.de> wrote:
> the error does not appear again. Only the error "Genome fasta headers contain whitespaces. Please reformat the headers" occured. I use ensembl genome fasta file (same as for PIGx RNA-seq), maybe I need to change it to the other file without whitespaces? What would you recommend? Thank you in advance.
>
> Best,
> Lisa
>
> Отправлено: 12 октября 2022 г. 14:22:59
> Кому: Kulaeva, Elizaveta
> Копия: pi...@googlegroups.com
> Тема: Re: [ext] Re: PIGx ChIP-seq pipeline error
>
> Looks like the indentation level of Peaks1/Peaks2 is wrong.
> They need to be indented such that it is a subfield under "peak_calling", which is also missing in your settings file.
>
> Check out `pigx chipseq --init` for full list and structure.
>
> Should look like
> ```
> general:
> params:
> ...
> peak_calling:
> Peak1:
> ....
> ```
>
>
> On Wed, Oct 12, 2022 at 2:17 PM Elizavet...@mdc-berlin.de <Elizavet...@mdc-berlin.de> wrote:
> Thank you very much, I apologise for such a silly mistake. Another (probably) silly question: how do I fix these mistakes? Peaks1 and peaks2 are calculations from two separate studies, each containing several control samples and several input samples.
> От: Bora Uyar <borauy...@gmail.com>
> Отправлено: 12 октября 2022 г. 14:05:59
> Кому: Kulaeva, Elizaveta
> Копия: pi...@googlegroups.com
> Тема: [ext] Re: PIGx ChIP-seq pipeline error
>
> Hi Elizaveta,
> In your bash script, you are trying to run pigx-rnaseq, that's why you get this error.
> Best,
> Bora
>
> On Wed, Oct 12, 2022 at 2:04 PM Elizavet...@mdc-berlin.de <Elizavet...@mdc-berlin.de> wrote:
> Dear PIGx ChIP-seq pipeline developers,
>
>
> I need to run PIGx to process the ChIP-seq data. I created files set_chipseq.yaml (for settings), sample_sheet_chipseq.csv, and a script test_chipseq.sh (I attach all these files to this email), and I got an error that the file cdna.fasta is missing, but this file type (cdna) is not needed for ChIP-seq: the tutorial stated that only the genome and the gtf file is needed.
>
> Here is the error message for the PIGx-ChIP-seq:
Elizavet...@mdc-berlin.de

And this is how, after applying formatting:

Отправлено: 12 октября 2022 г. 15:22:45
Кому: Kulaeva, Elizaveta
Копия: pi...@googlegroups.com; Bora Uyar
Alexander Blume
On 12. Oct 2022, at 15:45, Elizavet...@mdc-berlin.de wrote:
Thank you for your help. I have run these commands for fasta and gtf files but the error "Genome fasta headers contain whitespaces. Please reformat the headers" persists.Here is what the original headers look like in the fasta file:
<pastedImage.png>
And this is how, after applying formatting:
<pastedImage.png>
Elizavet...@mdc-berlin.de
Thank you. How long approximately should this line of code take to execute? I've already had it running for 30 minutes.
Lisa
Отправлено: 12 октября 2022 г. 15:48:34
Alexander Blume
Elizavet...@mdc-berlin.de
I tested this code:
Отправлено: 12 октября 2022 г. 16:58:58
Alexander Blume
cat Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa | awk '{ print $1}' > refGenome_noWhiteSpace.fa
To view this discussion on the web visit https://groups.google.com/d/msgid/pigx/9561e622d89d4c659e4fd4db85aba32c%40mdc-berlin.de.
Elizavet...@mdc-berlin.de
I just waited and it worked :) now I've started the pipeline, so far it's going without errors. Thank you very much for your help!
Отправлено: 12 октября 2022 г. 17:22:20
Alexander Blume
To view this discussion on the web visit https://groups.google.com/d/msgid/pigx/fff4bbe9c2d64028aa803272da9c3d50%40mdc-berlin.de.
Alexander Blume
Elizavet...@mdc-berlin.de
Hello, the ChIP-seq Pipeline has an error again (extract_nucleotide_frequency rule), I am sending you an error message (log file for this rule is empty):
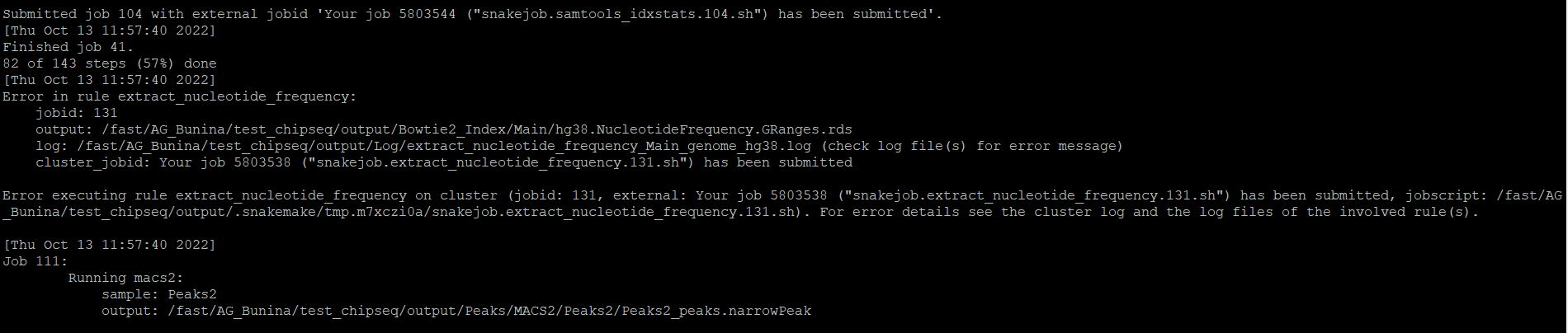
Thank you in advance,
Lisa
Отправлено: 12 октября 2022 г. 17:40
Alexander Blume
I would assume that your jobs requested resources were exceeded.
You have to check the associated log files in the Log/Cluster dir and may also check the status of the associated job using qacct -j <yourjobid>.
My recommended fix would be to just double the requested default memory in the settings file at
execution: rules: __default__: memory
On 13. Oct 2022, at 12:37, Elizavet...@mdc-berlin.de wrote:
Hello, the ChIP-seq Pipeline has an error again (extract_nucleotide_frequency rule), I am sending you an error message (log file for this rule is empty):
<pastedImage.png>
Elizavet...@mdc-berlin.de
Thank you, the problem was indeed related to the required memory.
Best,
Lisa
Отправлено: 13 октября 2022 г. 12:53:28