GMGC marine database indexing runs out of memory
39 views
Skip to first unread message

Alyse Larkin
Feb 8, 2022, 6:37:49 PM2/8/22
to NGLess
Hello,
I am attemping to annotate some short read metagenomes using my university's HPC platform. To start, I decided to run just two metagenomes as a test run on a single CPU with 1.5 TB of memory. My hope was to complete the indexing of the GMGC marine database so that I could then annotate the rest of my metagenome files on a lower memory node. However, even with 1.5 TB, I am still getting an out of memory error message.
My question is, should I expect the indexing of the GMGC marine database to take more than 1.5 TB of memory? Or do you suspect something else is going wrong here?
I have attached my .ngl script, my HPC submission script (.sub), and the error file (.err) with the command output with the associated error message here.
Thank you very much for your assistance!
Sincerely,
Alyse

Luis Pedro Coelho
Feb 9, 2022, 1:05:20 PM2/9/22
to Alyse Larkin, NGLess List
Hi Alyse,
Thanks for reaching out.
This is surprising to me. I think we have indexed the marine subset with much less memory than 1.5TB. Are you sure that the whole memory is being reserved for your job?
Best
Luis
Luis Pedro Coelho | Fudan University | http://luispedro.org
--You received this message because you are subscribed to the Google Groups "NGLess" group.To unsubscribe from this group and stop receiving emails from it, send an email to ngless+un...@googlegroups.com.To view this discussion on the web visit https://groups.google.com/d/msgid/ngless/1fda28a4-67e4-44b6-84d6-285f5d711c67n%40googlegroups.com.Attachments:
- ngless-single.sub
- gmgc_annotate_amt.ngl
- AMT1_Annotate-10180898.err
Alyse Larkin
Feb 9, 2022, 3:19:49 PM2/9/22
to NGLess
Hi Luis,
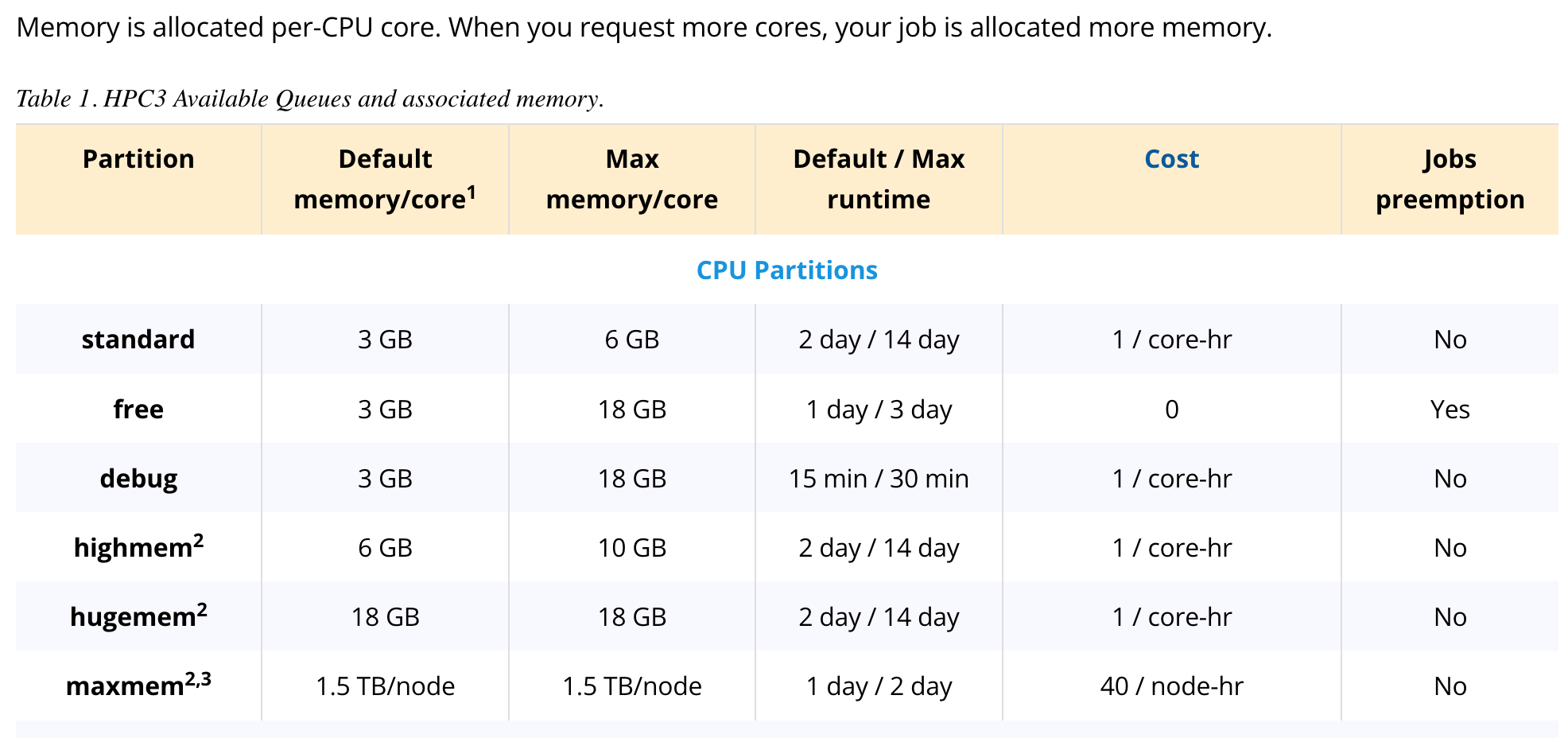
That's a good question! I thought the set up was 1.5 TB per core, but when I looked at our HPC partition structure again, it actually seems like it's 1.5 TB per node. Thus, it is likely that not all the memory is being reserved for the single core that I requested. (See screenshot of the set up below, I used the 'maxmem' partition)
However, I also ran the job on a core with a specific allocation of 18G of memory (i.e., the 'hugemem' partition) and got the same "out of memory" error at an earlier step in the indexing process. As such, I wanted to ask whether you have a sense of how much memory indexing the marine subset might require? If I have an approximate memory requirement to shoot for, I think I can specifically ask for that allocation on the 'maxmem' partition.
Thank you so much for your assistance!
All the best,
Alyse
Alyse Larkin
Feb 17, 2022, 1:10:42 PM2/17/22
to NGLess
Hello,
I am still struggling with the memory requirements for annotating metagenomes with the GMGC marine database. I successfully completed the indexing of both the gmgc:marine and the gmgc:marine:no-rare databases using a single thread with 100 GB of memory. However, when I tried to annotate my first metagenome I have so far run out of memory using every setting that I have tried.
Currently, I am using the following settings on our HPC:
12 CPUS at 18G memory per CPU
10G of scratch space for temporary files
To save memory, I am using the following options in ngless:
reference='gmgc:marine:no-rare'
block_size_megabases=25000
However, I am still receiving the following error from the server: "slurmstepd: error: Detected 1 oom-kill event(s) in step 10417860.batch cgroup. Some of your processes may have been killed by the cgroup out-of-memory handler."
I know that it is my job that is killing the memory (rather than other jobs on the same node) because I can check the max memory used and see that it reaches my maximum allotment (216G):
CPUTime MaxRSS
---------- ----------
3-03:14:00 212544864K
I have attached my .ngl script, my HPC submission script (.sub), and my command line output/error (.err) file.
If you are able to provide any guidance on how to resolve the memory issue I would greatly appreciate it. I am currently at a bit of a loss as to how to move forward. Thank you very much for your assistance!
Best regards,
Alyse
You received this message because you are subscribed to a topic in the Google Groups "NGLess" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/ngless/Bny6Y_GFJLs/unsubscribe.
To unsubscribe from this group and all its topics, send an email to ngless+un...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/ngless/1207bcb9-7933-4bf2-bc3a-e7129dda8e3dn%40googlegroups.com.
Alyse Larkin
Feb 17, 2022, 3:31:27 PM2/17/22
to NGLess
Just a quick update - I tried running the script with the "--subsample" flag just to ensure that the script would finish, but it still ran out of memory.
(base) [larkinsa@login-i15:/pub/larkinsa] $sacct -j 10514370 --format="CPUTime,MaxRSS"
CPUTime MaxRSS
---------- ----------
10:16:12 212433712K
I attached the error file. In this case, it looks like it is running out of memory at the subsample step. Does ngless load the metagenome that is being processed into memory? Even if that is the case, the files that I am processing are about 2G total (see below), which I wouldn't expect to take up the whole 216G of memory or even 10G of scratch space.
-rw-rw-r-- 1 larkinsa 856M Aug 26 2019 /dfs3b/BGS_Trimmed/AMT_Trimmed/paired_AMT2018_001_meta_R1.fastq.gz
-rw-rw-r-- 1 larkinsa 907M Aug 26 2019 /dfs3b/BGS_Trimmed/AMT_Trimmed/paired_AMT2018_001_meta_R2.fastq.gz
-rw-rw-r-- 1 larkinsa 253M Sep 2 2019 /dfs3b/BGS_Trimmed/AMT_Trimmed/unpaired_AMT2018_001_meta.fastq.gz
So in sum, ngless runs out of 216G of memory after about six hours if annotating a single genome with the gmgc:marine:no-rare database *or* it runs out of memory after 50 minutes if I add the --subsample flag.
Any thoughts? Thanks so much for your assistance!
All the best,
Alyse
Reply all
Reply to author
Forward
0 new messages