Taxonomic/functional annotations and interpretation of read abundance
176 views
Skip to first unread message

Quinten Ducarmon
Dec 17, 2019, 10:58:53 AM12/17/19
to NGLess
Dear,
I have run the NG-meta-profiler with NG-less version 1.0.1 (human-gut-profiler.ngl) on a simulated sample (only 1 sample to start with) of a small mock community. I have done this exactly according to the tutorial on http://ngless.embl.de/ng-meta-profiler.html.
While the run went fine and I obtained output files, I am not sure how to proceed because of several reasons:
1) For taxonomic annotation I obtained a motus.counts.txt file, where 9 different Clusters are identified. However, there is no taxonomic annotation which allows me to interpret which genus/species belongs to which Cluster. In addition, I am not sure how I should interpret the read counts (as to perform downstream statistical analysis)).
2) For functional annotation I have the same problem, I get a nice file called eggNOG.traditional.counts.txt but there is no 'biological annotation' with gene names or anything. Here, I did find a file (igc.functional.map) which was located in another folder with functional annotation. However, there are no matching names in the two files, so I cannot really proceed with this.
Hope you can help me out!
Quinten

Luis Pedro Coelho
Dec 17, 2019, 12:08:54 PM12/17/19
to Quinten Ducarmon, NGLess List
Hi Quinten,
A few quick answers (from memory, but I can hopefully still remember these details).
While the run went fine and I obtained output files, I am not sure how to proceed because of several reasons:
OK.
1) For taxonomic annotation I obtained a motus.counts.txt file, where 9 different Clusters are identified. However, there is no taxonomic annotation which allows me to interpret which genus/species belongs to which Cluster. In addition, I am not sure how I should interpret the read counts (as to perform downstream statistical analysis)).
These are motus clusters, you can get the information for them from the motus website https://motu-tool.org/. They are read counts. Normally, you want to do some normalization to take into account the differential gene depths
To be frank, this is already a bit out of date as we should update to motus 2, which came out earlier this year (basically, at the time we wrote the paper, motus1 was still the current version, but then both papers came out around the same time).
We do support it within ngless, but it has a slightly different interface:
2) For functional annotation I have the same problem, I get a nice file called eggNOG.traditional.counts.txt but there is no 'biological annotation' with gene names or anything. Here, I did find a file (igc.functional.map) which was located in another folder with functional annotation. However, there are no matching names in the two files, so I cannot really proceed with this.
We can actually provide slightly different things.
It is always based on eggnog and using eggnog-mapper annotated gene catalogs. The default, IIRC, is to provide eggnog orthologous group counts, which is what you are seeing.
Best,
Luis
Hope you can help me out!Quinten
--You received this message because you are subscribed to the Google Groups "NGLess" group.To unsubscribe from this group and stop receiving emails from it, send an email to ngless+un...@googlegroups.com.To view this discussion on the web visit https://groups.google.com/d/msgid/ngless/4c4766c8-615c-4d82-9cab-4e525707d37a%40googlegroups.com.
Quinten Ducarmon
Dec 18, 2019, 4:27:58 AM12/18/19
to NGLess
Hi Luis,
Op dinsdag 17 december 2019 18:08:54 UTC+1 schreef Luis Pedro Coelho:
Thank you very much for your quick reply.
For now, let me just focus on completely understanding how to process the taxonomic output, although later I will probably still bother you with some questions about the functional assignment if that is okay.
- Considering that mOTUs are based on single copy marker genes, actually no further normalization (apart from accounting for different sequencing depth between samples) is necessary right? So it would be ideal to export the count data table and then depending on which downstream statistical methods I'd use, use different normalization procedures (as method use different types of normalization, e.g. median ratio normalization, clr transformation, total sum scaling etc.?)
- While I found on the website that "Map between mOTUs v1 and mOTUs v2 clusters can be downloaded", this does still not contain any taxonomic assignment (for example, which species/total taxonomic assignment belongs to ref_mOTU_v2_3996)? Is there a separate mapping file for this that I could download somewhere?
- Any idea when the update will include mOTUs2? :). For now, would you suggest I continue with the motus2 wrapper in NG-less or rather switch to the tool itself? I don't have much experience processing raw data of metagenomes, so would very much appreciate your suggestions here. The disadvantage I am thinking of when using mOTUs2 is that there is no integrated filtering of reads based on qc etc.
- Lastly, perhaps a dumb question, but can mOTUs2 actually also detect fungi/small eukaryotes in the gut metagenomes? I am doubting about this since the marker genes were benchmarked on prokaryotic genomes (if I understood correctly). Or should I perhaps directly ask this question to one of the mOTUs2 developers stated on the website to contact?
Kind regards and your help is very much appreciated,
Quinten
Op dinsdag 17 december 2019 18:08:54 UTC+1 schreef Luis Pedro Coelho:
To unsubscribe from this group and stop receiving emails from it, send an email to ngl...@googlegroups.com.

Renato Alves
Dec 18, 2019, 5:40:41 AM12/18/19
to ngl...@googlegroups.com
Hi Quinten,
> - While I found on the website that "Map between mOTUs v1 and mOTUs v2
> clusters can be downloaded", this does still not contain any taxonomic
> assignment (for example, which species/total taxonomic assignment
> belongs to ref_mOTU_v2_3996)? Is there a separate mapping file for this
> that I could download somewhere?
The taxonomic annotation is available in the db_mOTU/db_mOTU_taxonomy*
files included with the mOTUs v2 tool.
> - Any idea when the update will include mOTUs2? :). For now, would you
> suggest I continue with the motus2 wrapper in NG-less or rather switch
> to the tool itself? I don't have much experience processing raw data of
> metagenomes, so would very much appreciate your suggestions here. The
> disadvantage I am thinking of when using mOTUs2 is that there is no
> integrated filtering of reads based on qc etc.
Like Luis mentioned the motus() ngless-contrib function is slightly
different from the v1 equivalent. That said, you should be able to adapt
the example
https://github.com/ngless-toolkit/ngless-contrib/blob/master/motus.ngm/README.md#example
. In this case reads are still QC'ed and preprocessed with ngless and
only after passed to mOTUs.
> - Lastly, perhaps a dumb question, but can mOTUs2 actually also detect
> fungi/small eukaryotes in the gut metagenomes? I am doubting about this
> since the marker genes were benchmarked on prokaryotic genomes (if I
> understood correctly). Or should I perhaps directly ask this question to
> one of the mOTUs2 developers stated on the website to contact?
Not as of version 2.5 . I believe there are plans to address this but
the universality of the marker genes and genomic features of eukaryotes
make the task more challenging.
You could try MetaPhlan2 which can provide some result. We also have an
ngless interface for it. The example
https://github.com/ngless-toolkit/ngless-contrib/blob/master/metaphlan.ngm/README.md#example
should be similar to the motus() one above.
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
> - While I found on the website that "Map between mOTUs v1 and mOTUs v2
> clusters can be downloaded", this does still not contain any taxonomic
> assignment (for example, which species/total taxonomic assignment
> belongs to ref_mOTU_v2_3996)? Is there a separate mapping file for this
> that I could download somewhere?
files included with the mOTUs v2 tool.
> - Any idea when the update will include mOTUs2? :). For now, would you
> suggest I continue with the motus2 wrapper in NG-less or rather switch
> to the tool itself? I don't have much experience processing raw data of
> metagenomes, so would very much appreciate your suggestions here. The
> disadvantage I am thinking of when using mOTUs2 is that there is no
> integrated filtering of reads based on qc etc.
different from the v1 equivalent. That said, you should be able to adapt
the example
https://github.com/ngless-toolkit/ngless-contrib/blob/master/motus.ngm/README.md#example
. In this case reads are still QC'ed and preprocessed with ngless and
only after passed to mOTUs.
> - Lastly, perhaps a dumb question, but can mOTUs2 actually also detect
> fungi/small eukaryotes in the gut metagenomes? I am doubting about this
> since the marker genes were benchmarked on prokaryotic genomes (if I
> understood correctly). Or should I perhaps directly ask this question to
> one of the mOTUs2 developers stated on the website to contact?
the universality of the marker genes and genomic features of eukaryotes
make the task more challenging.
You could try MetaPhlan2 which can provide some result. We also have an
ngless interface for it. The example
https://github.com/ngless-toolkit/ngless-contrib/blob/master/metaphlan.ngm/README.md#example
should be similar to the motus() one above.
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
Quinten Ducarmon
Dec 18, 2019, 6:08:06 AM12/18/19
to NGLess
Hi Renato,
Thanks a lot for this information, I will get to work with these tips!
One last question, was I right in my understanding of:
One last question, was I right in my understanding of:
"- Considering that mOTUs are based on single copy marker genes, actually no further normalization (apart from accounting for different sequencing depth between samples) is necessary right? So it would be ideal to export the count data table and then depending on which downstream statistical methods I'd use, use different normalization procedures (as method use different types of normalization, e.g. median ratio normalization, total sum scaling etc.?)"
Best and your quick reply is very much appreciated,
Quinten
Op woensdag 18 december 2019 11:40:41 UTC+1 schreef renato.alves:
Op woensdag 18 december 2019 11:40:41 UTC+1 schreef renato.alves:
Renato Alves
Dec 18, 2019, 6:50:19 AM12/18/19
to ngl...@googlegroups.com
> "- Considering that mOTUs are based on single copy marker genes,
> actually no further normalization (apart from accounting for different
> sequencing depth between samples) is necessary right? So it would be
> ideal to export the count data table and then depending on which
> downstream statistical methods I'd use, use different normalization
> procedures (as method use different types of normalization, e.g. median
> ratio normalization, total sum scaling etc.?)"
Yes.
> actually no further normalization (apart from accounting for different
> sequencing depth between samples) is necessary right? So it would be
> ideal to export the count data table and then depending on which
> downstream statistical methods I'd use, use different normalization
> procedures (as method use different types of normalization, e.g. median
> ratio normalization, total sum scaling etc.?)"
You can also ask for the output to be in relative abundance instead of
counts. In the example I linked before this is what is being requested.
If you want counts only you can simply omit the
"relative_abundance=true" line.
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
orcid.org/0000-0002-7212-0234
havior.
Quinten Ducarmon
Dec 18, 2019, 9:22:13 AM12/18/19
to NGLess
Hi Renato,
Sorry that I keep spamming you, but I'm experiencing some troubles with the motu2 wrapper in ngless. Probably due to my limited knowledge of these issues and especially ngless, but I hope you can help me.
I have cloned the repository (all modules) and ran the install.sh script. In addition, I have loaded the ngless conda environment. So this all seems fine
However, I do not know how I get the motus function to work (run) as indicated in https://github.com/ngless-toolkit/ngless-contrib/tree/master/motus.ngm. Perhaps you could provide an example line of code for the motus function which would process a sample?
I also tried changing the motu version from 0.1 to 2.5 in the human-gut-profiler.ngl, but then the ng meta profiler does not work anymore (not sure if this made any sense anyway).
Best,
Quinten
Op woensdag 18 december 2019 12:50:19 UTC+1 schreef renato.alves:
Op woensdag 18 december 2019 12:50:19 UTC+1 schreef renato.alves:
Email: renat...@embl.de
orcid.org/0000-0002-7212-0234
havior.
Renato Alves
Dec 18, 2019, 11:36:58 AM12/18/19
to ngl...@googlegroups.com
Hi Quinten,
> Sorry that I keep spamming you, but I'm experiencing some troubles with
> the motu2 wrapper in ngless. Probably due to my limited knowledge of
> these issues and especially ngless, but I hope you can help me.
Can you specify what have you tried and what didn't work?
Did you get any error?
> I have cloned the repository (all modules) and ran the install.sh
> script. In addition, I have loaded the ngless conda environment. So this
> all seems fine
> However, I do not know how I get the motus function to work (run) as
> indicated
> in https://github.com/ngless-toolkit/ngless-contrib/tree/master/motus.ngm.
> Perhaps you could provide an example line of code for the motus function
> which would process a sample?
The readme I linked before includes an example at the end
(https://github.com/ngless-toolkit/ngless-contrib/tree/master/motus.ngm#example).
This is a complete script.
Given some samples located in the current folder and their name added to
a text file "all_samples.txt" (one per line). Running the script through
ngless should produce an output file "all_samples.motusv2.relabund.tsv".
You will need to run ngless as many times as the number of samples.
> I also tried changing the motu version from 0.1 to 2.5 in the
> human-gut-profiler.ngl, but then the ng meta profiler does not work
> anymore (not sure if this made any sense anyway).
Yes, motus 2.5 is an external ngless module contributed by the ngless
community. As Luis mentioned before, it behaves differently and being
external needs to be loaded with the "local" prefix and installed manually.
You can find additional information on how external modules work in the
official documentation: https://ngless.readthedocs.io/en/latest/modules.html
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
> Sorry that I keep spamming you, but I'm experiencing some troubles with
> the motu2 wrapper in ngless. Probably due to my limited knowledge of
> these issues and especially ngless, but I hope you can help me.
Did you get any error?
> I have cloned the repository (all modules) and ran the install.sh
> script. In addition, I have loaded the ngless conda environment. So this
> all seems fine
> However, I do not know how I get the motus function to work (run) as
> indicated
> in https://github.com/ngless-toolkit/ngless-contrib/tree/master/motus.ngm.
> Perhaps you could provide an example line of code for the motus function
> which would process a sample?
(https://github.com/ngless-toolkit/ngless-contrib/tree/master/motus.ngm#example).
This is a complete script.
Given some samples located in the current folder and their name added to
a text file "all_samples.txt" (one per line). Running the script through
ngless should produce an output file "all_samples.motusv2.relabund.tsv".
You will need to run ngless as many times as the number of samples.
> I also tried changing the motu version from 0.1 to 2.5 in the
> human-gut-profiler.ngl, but then the ng meta profiler does not work
> anymore (not sure if this made any sense anyway).
community. As Luis mentioned before, it behaves differently and being
external needs to be loaded with the "local" prefix and installed manually.
You can find additional information on how external modules work in the
official documentation: https://ngless.readthedocs.io/en/latest/modules.html
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
orcid.org/0000-0002-7212-0234
Message has been deleted
Quinten Ducarmon
Dec 19, 2019, 5:34:11 AM12/19/19
to NGLess
Hi Renato,
Op woensdag 18 december 2019 17:36:58 UTC+1 schreef renato.alves:
Thanks again for the quick answer. I think I will try to just run the mOTUs2 separately from the NG-meta-profiler as I am getting quite confused.
So I understand that I have to change my human-gut-profiler.ngl file and integrate the changes as described here. https://github.com/ngless-toolkit/ngless-contrib/blob/master/motus.ngm/README.md#example. However, this is where it becomes too confusing for me. Obviously I can include the "local" prefix for motus import, but how to integrate the rest of the script is a bit elusive to me. For example, which part of the human-gut-profiler.ngl file do I take out (especially regarding the motus1 part at the end of the script) and how to integrate the rest of the script, considering that in https://github.com/ngless-toolkit/ngless-contrib/blob/master/motus.ngm/README.md#example suddenly the inputs become 'sample' and 'files' which was not the case in human-gut-profiler.ngl. In there, the specI_reads were used as input for creating the motus.
I know this is not the most useful feedback from my side, but I just don't really understand how to properly adapt the script to get the wrapper working.
So I guess the best for me would be to continue with the mOTUs2 separately and wait for the update where mOTUs 2.5 is integrated in NG-meta-profiler!
Best,
Quinten
Op woensdag 18 december 2019 17:36:58 UTC+1 schreef renato.alves:
Renato Alves
Dec 19, 2019, 6:29:17 AM12/19/19
to ngl...@googlegroups.com
Hi Quinten,
Can you check
https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/extra/human-gut-profiler-motus-v2.ngl
This still requires validation but produces results using motus-v2.5.
Note though that due to differences in how v1 and v2 were implemented,
this script produces only one "motus" file per sample.
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
Can you check
https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/extra/human-gut-profiler-motus-v2.ngl
This still requires validation but produces results using motus-v2.5.
Note though that due to differences in how v1 and v2 were implemented,
this script produces only one "motus" file per sample.
Cheers,
Renato
--
Renato Alves, PhD
Bork Group
EMBL-Heidelberg, Germany
Lab Phone: +49 6221 387 8150
orcid.org/0000-0002-7212-0234
Renato Alves
Dec 19, 2019, 7:30:38 AM12/19/19
to ngl...@googlegroups.com
> Oh thanks a lot, I wasn't aware of that script. I will have a look and
> let you know if I can get it to work, and whether it produces the
> desired output with my simulated data!
It didn't exist yesterday :) which is why I said it needs validation.
Let me know how it goes and if you run into any problems.
Also, if you encounter errors, please pass the option --trace to ngless
so we get additional output that will help debugging possible issues.
> let you know if I can get it to work, and whether it produces the
> desired output with my simulated data!
It didn't exist yesterday :) which is why I said it needs validation.
Let me know how it goes and if you run into any problems.
Also, if you encounter errors, please pass the option --trace to ngless
so we get additional output that will help debugging possible issues.
Quinten Ducarmon
Dec 20, 2019, 7:46:59 AM12/20/19
to NGLess
Hi Renato,
First of all, very happy to let you know it works and produces the 2 output files it should produce.
The output is however quite a bit different from using the mOTUs2.5 directly.
With using the mOTUs 2.5 directly I correctly obtained 8 different bacterial species (which is what is contained in my simulated mock community, the bacteria in there are the ones contained in https://files.zymoresearch.com/protocols/_d6300_zymobiomics_microbial_community_standard.pdf). They were not all correctly classified up until species level, but the good thing is that I only got 8 mOTUs with reads with approximately similar abundance (which is what is expected).
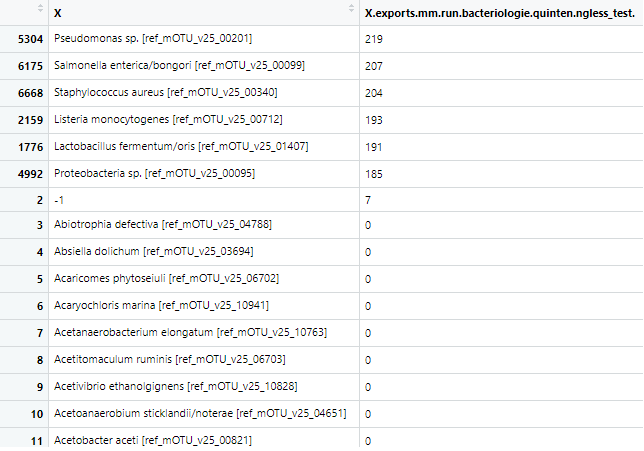
However, with NG-meta-profiler I only obtained 6 of the species and with much lower read counts (~700 using mOTUs 2.5 directly, ~200 using NG-meta-profiler). I am not dectecting Bacillus subtilis and Enterococcus faecalis for some reason. Interestingly, these were the species that could not be accurately classified using the mOTUs 2.5 tool directly. Could they somehow be filtered out in the ng-meta-profiler? See attached part of the output sorted on read abundance for both runs, that should explain a bit better.
So I guess it is a bit unexpected that due to the filtering of human reads and low quality reads (there shouldn't be any human reads in the simulated data, and all reads should be high quality) the output is quite different? Any idea what could be happening here?
In any case, I am going on holiday from tomorrow onwards until the first week of January, so then I will definitely be back with some questions =)
Thanks a lot for your help, I wish you a merry Christmas and the best wishes for the New Year!
Quinten
Op donderdag 19 december 2019 13:30:38 UTC+1 schreef renato.alves:
Op donderdag 19 december 2019 13:30:38 UTC+1 schreef renato.alves:
Quinten Ducarmon
Jan 14, 2020, 8:43:19 AM1/14/20
to NGLess
Hi Renato,
Did you have any time yet to look at my remark/question on why the output seems to be worse for some reason?
Your help is much appreciated, as always.
Best,
Quinten
Op vr 20 dec. 2019 om 13:47 schreef Quinten Ducarmon <quinten...@gmail.com>:
--
You received this message because you are subscribed to the Google Groups "NGLess" group.
To unsubscribe from this group and stop receiving emails from it, send an email to ngless+un...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/ngless/f434627e-8032-4448-8fcb-aef1fb28019d%40googlegroups.com.
Renato Alves
Jan 14, 2020, 9:54:25 AM1/14/20
to ngl...@googlegroups.com
-----BEGIN PGP SIGNED MESSAGE-----
Hash: SHA256
Hi Quinten,
If I understand your question, you are asking why the results from mOTUs 2.5 are different from, and better than, those produced with NG-meta-profiler.
The reason is that NG-meta-profiler [1] uses an algorithm that is equivalent to mOTUs 1.0.
Consequently it is expectable that results from mOTUs 2.5 are better than mOTUs 1.0 simply because the available data at the time of the construction of each of these tools was considerably different.
Perhaps this [2] illustration will help clarify the differences.
If by NG-meta-profiler, in fact, you are referring to results produced with [3], then I can't say for sure.
The only difference between [3] and using mOTUs 2.5 directly is that the former removes reads that map to the human genome (i.e. human contamination).
It could be that reads from some species are similar to regions in the human genome but this is an unlikely case.
The alternative of human regions being present in some bacterial reference genomes is a known occurrence but unlikely to have a significant effect here considering how the underlying mOTUs database is constructed and used.
Hope this helps,
Renato
[1] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/human-gut-profiler.ngl
[2] https://raw.githubusercontent.com/motu-tool/mOTUs_v2/master/pics/mOTUs_versions.png
[3] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/extra/human-gut-profiler-motus-v2.ngl
- --
Renato Alves, Dr. rer. nat.
EMBL Bio-IT & HD-HuB (ELIXIR Germany)
EMBL - Meyerhofstr. 1, 69117 Heidelberg, Germany
Office Phone: +49 6221 387-8953
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
Github/Gitlab: @unode - Twitter: @renato_alvs
-----BEGIN PGP SIGNATURE-----
iQIzBAEBCAAdFiEEqLl/Wmidk+Oyh+vw8TuUyrVegSsFAl4d1h8ACgkQ8TuUyrVe
gSsT0A/8DlrWzqBF4wPnMHh2naotNCnfRAMXGmKSNIdc2tcXk0YLJfDRbsalxfGP
SUX9BbOjbzi3oKyc75R0lmNedR6074DESPxoeqDL/4EshqZh00F/Ucn/N5jwz2QJ
R+di9/nIw+OaO8IqWty+NuIy22B2/FkI9fFox/slAJr7QoWVOnkfL7l4grc+2KQv
JlzoCNgIYsWo6pLw8WemaK+eKK/kErdRSF/cd4cdr04LIbUmmlRRccRsroYdGRG/
zMF+9P9Xa17J9kZ5jABxVyBsLnn6PDzXr6rW7NmTOnGt39m5mdboc+WIEAHo+OS/
mN33wPEb/FdQtoFuIdEqkW5uuuu4tOeScYIumLM3xGkrQTJYRIovEXIuKaf7HC1p
fLaxvUWkMJxziICB0IG2HrphbzeDUmJurkioDvg6A3uYmbRVj8nnDSyR4ytINZlc
qNTKMfUQx0FDQmnnis+CLOjJ/uQfQnTch6Rsx3yX73oew8joeFg2WFR/R/ktgFp5
ojVb5qs1y3nc1bP6Xa8BvuaJp96PPB4zN1CEcktL+Zbuvc5oJl28DVwzUJKi2bz9
bqwUxKh2EdS5b4gRwJGiRFmKdI5qIxitYb1RfM626tK1PLv7pjYcsosd/NtLwBDS
dri/D/KrxBMtKZNaHHnAkuKXmdlNhJl3WDP7soTXi4YYRJPbanM=
=h699
-----END PGP SIGNATURE-----
Hash: SHA256
Hi Quinten,
If I understand your question, you are asking why the results from mOTUs 2.5 are different from, and better than, those produced with NG-meta-profiler.
The reason is that NG-meta-profiler [1] uses an algorithm that is equivalent to mOTUs 1.0.
Consequently it is expectable that results from mOTUs 2.5 are better than mOTUs 1.0 simply because the available data at the time of the construction of each of these tools was considerably different.
Perhaps this [2] illustration will help clarify the differences.
If by NG-meta-profiler, in fact, you are referring to results produced with [3], then I can't say for sure.
The only difference between [3] and using mOTUs 2.5 directly is that the former removes reads that map to the human genome (i.e. human contamination).
It could be that reads from some species are similar to regions in the human genome but this is an unlikely case.
The alternative of human regions being present in some bacterial reference genomes is a known occurrence but unlikely to have a significant effect here considering how the underlying mOTUs database is constructed and used.
Hope this helps,
Renato
[1] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/human-gut-profiler.ngl
[2] https://raw.githubusercontent.com/motu-tool/mOTUs_v2/master/pics/mOTUs_versions.png
[3] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/extra/human-gut-profiler-motus-v2.ngl
- --
Renato Alves, Dr. rer. nat.
EMBL Bio-IT & HD-HuB (ELIXIR Germany)
EMBL - Meyerhofstr. 1, 69117 Heidelberg, Germany
Office Phone: +49 6221 387-8953
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
Github/Gitlab: @unode - Twitter: @renato_alvs
-----BEGIN PGP SIGNATURE-----
iQIzBAEBCAAdFiEEqLl/Wmidk+Oyh+vw8TuUyrVegSsFAl4d1h8ACgkQ8TuUyrVe
gSsT0A/8DlrWzqBF4wPnMHh2naotNCnfRAMXGmKSNIdc2tcXk0YLJfDRbsalxfGP
SUX9BbOjbzi3oKyc75R0lmNedR6074DESPxoeqDL/4EshqZh00F/Ucn/N5jwz2QJ
R+di9/nIw+OaO8IqWty+NuIy22B2/FkI9fFox/slAJr7QoWVOnkfL7l4grc+2KQv
JlzoCNgIYsWo6pLw8WemaK+eKK/kErdRSF/cd4cdr04LIbUmmlRRccRsroYdGRG/
zMF+9P9Xa17J9kZ5jABxVyBsLnn6PDzXr6rW7NmTOnGt39m5mdboc+WIEAHo+OS/
mN33wPEb/FdQtoFuIdEqkW5uuuu4tOeScYIumLM3xGkrQTJYRIovEXIuKaf7HC1p
fLaxvUWkMJxziICB0IG2HrphbzeDUmJurkioDvg6A3uYmbRVj8nnDSyR4ytINZlc
qNTKMfUQx0FDQmnnis+CLOjJ/uQfQnTch6Rsx3yX73oew8joeFg2WFR/R/ktgFp5
ojVb5qs1y3nc1bP6Xa8BvuaJp96PPB4zN1CEcktL+Zbuvc5oJl28DVwzUJKi2bz9
bqwUxKh2EdS5b4gRwJGiRFmKdI5qIxitYb1RfM626tK1PLv7pjYcsosd/NtLwBDS
dri/D/KrxBMtKZNaHHnAkuKXmdlNhJl3WDP7soTXi4YYRJPbanM=
=h699
-----END PGP SIGNATURE-----
Renato Alves
Jan 15, 2020, 11:32:25 AM1/15/20
to ngless
-----BEGIN PGP SIGNED MESSAGE-----
Hash: SHA256
I spoke with one of the developers of mOTUs and he was equally surprised about this result.
Hash: SHA256
If you happen to get consistently worse results we'd be interested in figuring out why.
Let us know how this goes.
Cheers,
Renato
On 1/15/20 8:53 AM, Quinten Ducarmon wrote:
> Hi Renato,
>
> Thanks for your explanation.
> I was indeed referring to the latter (If by NG-meta-profiler, in fact, you are referring to results produced with [3], then I can't say for sure). Strange that the results are so different then indeed, I will have a bit deeper look and try the pipeline with some other data.
> I will also think about whether to use, if results for some reason remain a bit worse, to use mOTUs 2.5 separately (and then to somehow also do the eggNog separately).
>
> Thanks a lot for your feedback!
> Quinten
>
> Op di 14 jan. 2020 om 15:54 schreef Renato Alves <renato...@embl.de <mailto:renato...@embl.de>>:
>
> Hi Quinten,
>
> If I understand your question, you are asking why the results from mOTUs 2.5 are different from, and better than, those produced with NG-meta-profiler.
>
> The reason is that NG-meta-profiler [1] uses an algorithm that is equivalent to mOTUs 1.0.
> Consequently it is expectable that results from mOTUs 2.5 are better than mOTUs 1.0 simply because the available data at the time of the construction of each of these tools was considerably different.
> Perhaps this [2] illustration will help clarify the differences.
>
> If by NG-meta-profiler, in fact, you are referring to results produced with [3], then I can't say for sure.
> The only difference between [3] and using mOTUs 2.5 directly is that the former removes reads that map to the human genome (i.e. human contamination).
> It could be that reads from some species are similar to regions in the human genome but this is an unlikely case.
> The alternative of human regions being present in some bacterial reference genomes is a known occurrence but unlikely to have a significant effect here considering how the underlying mOTUs database is constructed and used.
>
> Hope this helps,
> Renato
>
>
> [1] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/human-gut-profiler.ngl
> [2] https://raw.githubusercontent.com/motu-tool/mOTUs_v2/master/pics/mOTUs_versions.png
> [3] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/extra/human-gut-profiler-motus-v2.ngl
>
>
> Hi Quinten,
>
> If I understand your question, you are asking why the results from mOTUs 2.5 are different from, and better than, those produced with NG-meta-profiler.
>
> The reason is that NG-meta-profiler [1] uses an algorithm that is equivalent to mOTUs 1.0.
> Consequently it is expectable that results from mOTUs 2.5 are better than mOTUs 1.0 simply because the available data at the time of the construction of each of these tools was considerably different.
> Perhaps this [2] illustration will help clarify the differences.
>
> If by NG-meta-profiler, in fact, you are referring to results produced with [3], then I can't say for sure.
> The only difference between [3] and using mOTUs 2.5 directly is that the former removes reads that map to the human genome (i.e. human contamination).
> It could be that reads from some species are similar to regions in the human genome but this is an unlikely case.
> The alternative of human regions being present in some bacterial reference genomes is a known occurrence but unlikely to have a significant effect here considering how the underlying mOTUs database is constructed and used.
>
> Hope this helps,
> Renato
>
>
> [1] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/human-gut-profiler.ngl
> [2] https://raw.githubusercontent.com/motu-tool/mOTUs_v2/master/pics/mOTUs_versions.png
> [3] https://github.com/ngless-toolkit/ng-meta-profiler/blob/master/extra/human-gut-profiler-motus-v2.ngl
>
>
> --
> You received this message because you are subscribed to the Google Groups "NGLess" group.
> To unsubscribe from this group and stop receiving emails from it, send an email to ngless+un...@googlegroups.com <mailto:ngless%2Bunsu...@googlegroups.com>.
> You received this message because you are subscribed to the Google Groups "NGLess" group.
> To view this discussion on the web visit https://groups.google.com/d/msgid/ngless/3b33e1e5-7f99-c1a5-7e67-a21174083196%40embl.de.
>
- --
Renato Alves, Dr. rer. nat.
EMBL Bio-IT & HD-HuB (ELIXIR Germany)
EMBL - Meyerhofstr. 1, 69117 Heidelberg, Germany
Office Phone: +49 6221 387-8953
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
Github/Gitlab: @unode - Twitter: @renato_alvs
-----BEGIN PGP SIGNATURE-----
iQIzBAEBCAAdFiEEqLl/Wmidk+Oyh+vw8TuUyrVegSsFAl4fPpcACgkQ8TuUyrVe
- --
Renato Alves, Dr. rer. nat.
EMBL Bio-IT & HD-HuB (ELIXIR Germany)
EMBL - Meyerhofstr. 1, 69117 Heidelberg, Germany
Office Phone: +49 6221 387-8953
Email: renato...@embl.de
orcid.org/0000-0002-7212-0234
Github/Gitlab: @unode - Twitter: @renato_alvs
-----BEGIN PGP SIGNATURE-----
gSsSrg//Q8zQzLpZtQPRzjZ2H13vtbV5JcuqBMdFd3NogFtki4lj2y5C1Dsamo5c
M0e563t7aQn0F5G1OX2CLTA9WATrOr9AZAwWVbGpvFx2w6q6dkcj2sa6gpc8TRld
sah9uztpGEmtM0uCmxVIwizn3yEyI31H4GWdrbFqeqbrLaODSZBPoSPjV3E32r2O
luO94wC4ISv67mr5c4Wh/xnmZFXpHMJJDmjZqNLLr3wRrDy7EpaU9FpIsDA4lvf5
mgSLnfEodASdfJFGkE1JNQ4h+kYF6qN+KmucYkDPFExKHjnF4JLAT9yM4m+hscDA
ujMGGbvBsiwptM2V5XGITn8St+L0VMO3k4Pvq0zqfAcczs2TPOuuTGaO00eNpo44
Q9vwc5C4Mmbib6WYAfbQGUebGWzeM4XkUBLEJ3tiF6zr1zqjOftq4XfPPsh46/w5
+stzZg1qyhxJp2LkD/103NoSjgLKlWAC1yb+CD9TUwM+Iku7KveJYAQC3PxH+hrK
DJnM5ZWHGQkEmmjaTy1Yu/xiYJXhc7iLEAV9Fq5+cL4+CD8W2a666XvreOGmCl8i
KOphZb8vSWtqdzTpgpxBhdALgio9QCUHFyaK4vywYqmng16//9D0lNq8ND+4kjYB
gE4E0briptHs2pN/Op+NcSsZ9qorSi8BvkGg7sNWT2/4DOUvQyE=
=HH8+
-----END PGP SIGNATURE-----

Ulrike Löber
Feb 20, 2020, 3:15:27 AM2/20/20
to NGLess
Hi all,
I would guess my question is somehow related to the question of Quinten. I ran the mouse-gut-profiler and got eggNOG.traditional.counts.txt , specI.raw.counts.txt , specI.scaled.counts.txt for each sample. All results are annotated as ClusterXYZ or 00009@acidNOG . How do I get meaningful annotation out of that, how do you usually combine the results and (as suggested I just used the script as it should be up to date) e.g. how do I know specI v3 was used ?
I would be very happy if you could give some more information about the "not-yet downstream" analysis. Are there any mapping files comming along with ngless I'm missing?
kind regards,
Ulrike
Quinten Ducarmon
Mar 30, 2020, 3:09:49 AM3/30/20
to Ulrike Löber, Renato Alves, NGLess
Hi Renato and Ulrike,
First of all a small question @Renato. I processed my samples for functional annotation using the KEGG database and normalized the output by gene length ({normed}) argument (everything worked very well and the ngless explanations were clear). My question is regarding the output. Is it acceptable (I'd say so, but just wanted to check), to scale the resulting output using total sum scaling, so that the samples are comparable?
@Ulrike, I found some downloads available on the eggnog website, but (at least to me) it was also not clear which file containing such mapping information.... http://eggnog5.embl.de/download/eggnog_5.0/. When annotating using KEGG KOs, the mapping file could be found a bit easier (https://www.genome.jp/kegg-bin/get_htext?ko00001), although I am currently working on getting all KEGG information in a nice workable table format.
Best,
Quinten
Op do 20 feb. 2020 om 09:15 schreef 'Ulrike Löber' via NGLess <ngl...@googlegroups.com>:
--
You received this message because you are subscribed to the Google Groups "NGLess" group.
To unsubscribe from this group and stop receiving emails from it, send an email to ngless+un...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/ngless/5155d0a3-a8af-49a7-a686-c9926e039246%40googlegroups.com.
Renato Alves
Apr 2, 2020, 7:17:28 PM4/2/20
to ngl...@googlegroups.com
-----BEGIN PGP SIGNED MESSAGE-----
Hash: SHA256
Hash: SHA256
> First of all a small question @Renato. I processed my samples for functional annotation using the KEGG database and normalized the output by gene length ({normed}) argument (everything worked very well and the ngless explanations were clear). My question is regarding the output. Is it acceptable (I'd say so, but just wanted to check), to scale the resulting output using total sum scaling, so that the samples are comparable?
Using TSS should be fine but depending what you are trying to compare you will need to decide how to handle the -1 fraction.
> @Ulrike, I found some downloads available on the eggnog website, but (at least to me) it was also not clear which file containing such mapping information.... http://eggnog5.embl.de/download/eggnog_5.0/. When annotating using KEGG KOs, the mapping file could be found a bit easier (https://www.genome.jp/kegg-bin/get_htext?ko00001), although I am currently working on getting all KEGG information in a nice workable table format.
Sorry for the brevity, very busy days...
Renato
-----BEGIN PGP SIGNATURE-----
iQIzBAEBCAAdFiEEqLl/Wmidk+Oyh+vw8TuUyrVegSsFAl6GcoYACgkQ8TuUyrVe
gStoAA//ee2yT3g4WS/nTBU/KqmPL7DsHZfhnRd4YL8nliQ1j4ITq/p5s/BMmcpG
UTAf7waUKqIoZwVsELPWU9GUKH6BzukN0BE1peCDkOWA2imfqp/ioULvv9mvVEuQ
aSwyiSEJa8sRm5R085EYaz9+J8bN0k7GgM1QM/hG0jsVp+tYrf9AfEWqNfUVzY7R
iXCkC0SGsvXSKyhfVD72nNFJc50kGYBlE2aNIzKW3LU4Py/6yVKdgOFqakZAFZhK
sIk6/xZu0e71Pul/XLapx3q3n01xsFXvIkI1uufYKTGlMKCrwY9Nez2v8SRt0K83
VoihZDlneCN6IeLy/NZ0VnAxBlJecTEUtOF5TSO05YQF6VWYS4rfF/J/MzH6jnpK
T2MFT2hS9Nas6bp3W0Y9RWEyviD37qI2UpwGXnwcHwKnSRGCkZDyucm504IdCcUp
E6ZG3OVsxaew3fEE249nQpAusXp8whzE02c5ihGcs4T6mbToXUPLAk7ZdmnRYiMp
gwHEaIMoeTqNttMDy3bLeOiCKphnz2fLr2YYWQ/kE3HMVqCwsP8KI/i1mXF5MhVJ
iLrg6e0kNO9Q1V+LP4LvT/OMUPNK/t5TNMsFSd8zXsfn4m9wJd8Aa7LxLDv+fM29
ACBDOG8e3Qk7mM1+RfxYy7gD2Cq7wFJmt2y5WB8uj4btJrRwGm0=
=XtP3
-----END PGP SIGNATURE-----

{kind=link}
{kind=link}
Christian Hodar
Jan 4, 2021, 8:48:59 AM1/4/21
to NGLess
Hi Queten,
I recently joined this group, and I read your question so I hope that my answer don't be too late.
About the KEGG orthology, I downloaded the json file available here (https://www.genome.jp/kegg-bin/download_htext?htext=ko00001&format=json&filedir=) and parse it to get a tabular form of the ontology.
I attach you my file.
All the best.
Christian.
Christian.
Reply all
Reply to author
Forward
0 new messages