problems in choosing nstates in SA-CASSCF calculation and MS-MR-CASPT2 calculation

Bin Han
memory,1000,m
direct
gprint,basis,orbital,civector
symmetry,nosym
geomtyp=xyz
geometry=R-MeN8-EuI2.xyz
***,caspt2
memory,1000,m
direct
gprint,basis,orbital,civector
symmetry,nosym
geomtyp=xyz
geometry=R-MeN8-EuI2.xyz
set,charge=2
include,svp.incl
cfit,basis=qzvpp/jkfit
{df-hf
occ,147
closed,140
wf,287,1,7}
{avas
start,2100.2
orbital,2110.2
center,81,5d,4f}
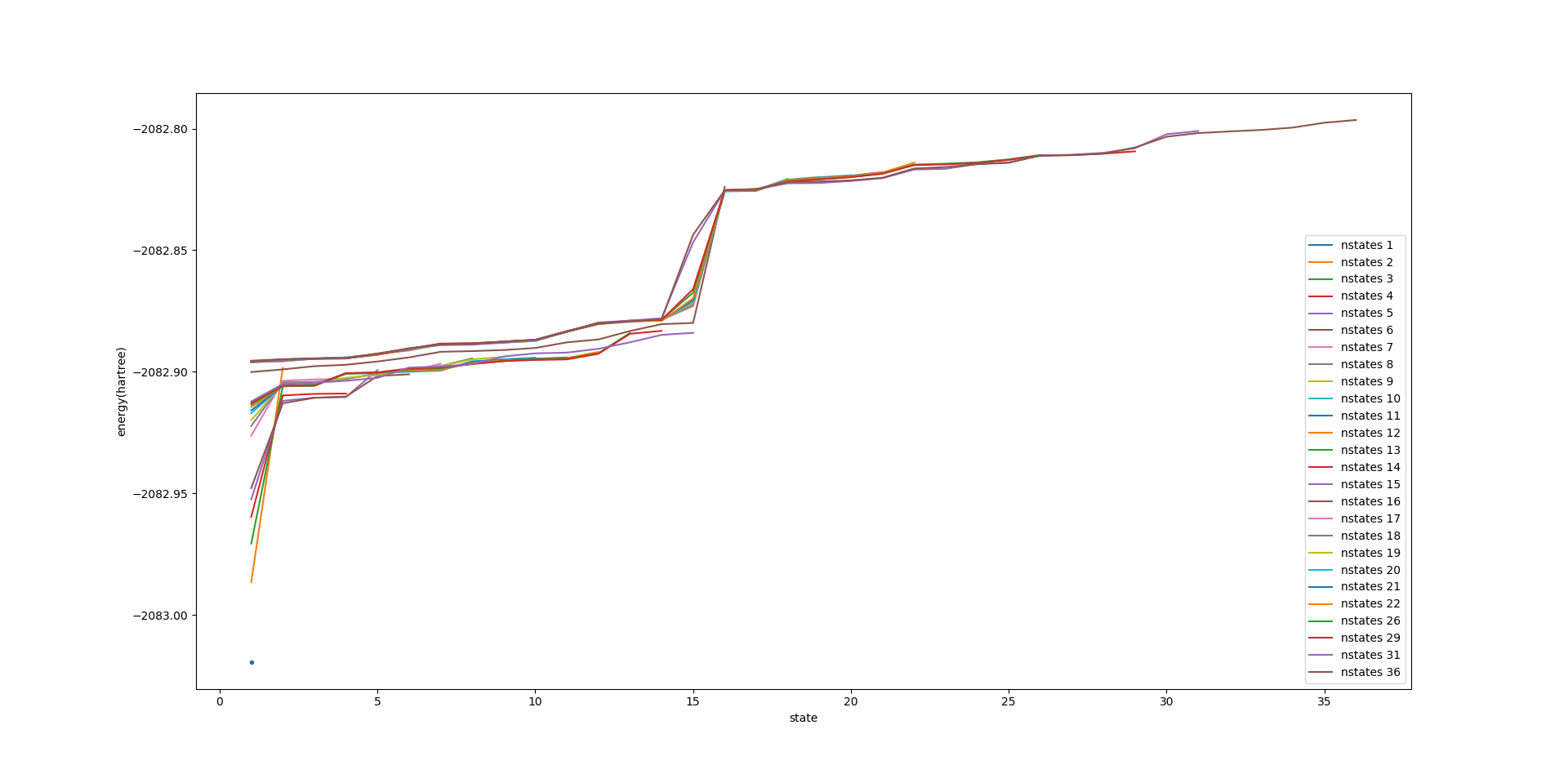
nstate=[1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]
i=0
do istate=1,#nstate
i=i+1
state(i)=nstate(istate)
{df-multi
closed,140
occ,152
wf,287,1,7
start,2110.2
state,state(i)
canonical}
multi(i)=energy(2)-energy(1)
enddo
{table,state,multi
head,state,multi}
memory,2000,m
direct
gprint,basis,orbital,civector
symmetry,nosym
geomtyp=xyz
geometry=R-MeN8-EuI2.xyz
set,charge=2
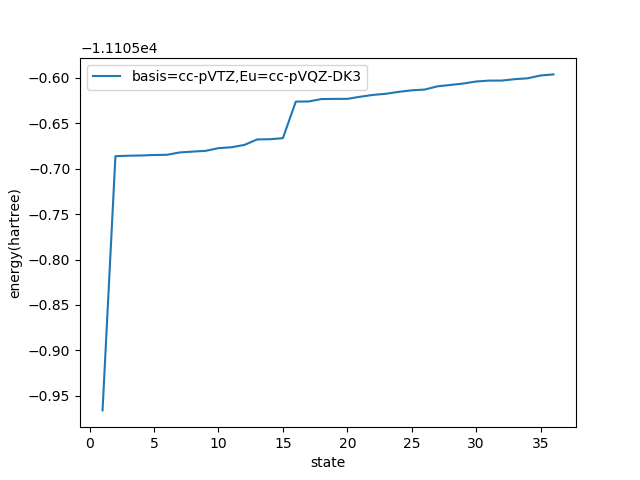
basis=cc-pVTZ,Eu=cc-pVQZ-DK3
{hf
closed,154
occ,161
wf,315,1,7
avas
center,81,4f,5d}
{multi,SO-SCI
closed,154
occ,166
wf,315,1,7
state,36
canonical}

{kind=link}
{kind=link}
Peterson, Kirk
Hi,
I can comment on the 2nd set of calculations. While you chose a relativistically contracted basis set for Eu, you didn't actually turn on the relativistic Hamiltonian. Please use cc-pVTZ-DK for the light atoms and specify dkho=3 before your first energy calculation.
regards,
-Kirk
From: <molpr...@googlegroups.com> on behalf of Bin Han <ice86...@gmail.com>
Date: Monday, June 6, 2022 at 5:54 AM
To: molpro-user <molpr...@googlegroups.com>
Subject: [molpro-user] problems in choosing nstates in SA-CASSCF calculation and MS-MR-CASPT2 calculation
[EXTERNAL EMAIL] DO NOT CLICK links or attachments unless you recognize the sender and know the content is safe.
--
You received this message because you are subscribed to the Google Groups "molpro-user" group.
To unsubscribe from this group and stop receiving emails from it, send an email to
molpro-user...@googlegroups.com.
To view this discussion on the web, visit
https://groups.google.com/d/msgid/molpro-user/192bf563-4027-42a3-bf15-d6ff66d3dfb2n%40googlegroups.com.
Bin Han
MAXITER,400
closed,140
occ,152
wf,287,1,7
start,2110.2
state,36;WEIGHT,1,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143
canonical}
{df-rs2c,shift=0.1,IPEA=0.3,mix=36,xms=1,noprop,maxit=100,maxiti=400
closed,140
occ,152
core,140
wf,287,1,7
state,36;WEIGHT,1,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143,0.02857143}
MAXITER,400
closed,140
occ,152
wf,287,1,7
start,2110.2
state,2
canonical}
{rs2c,shift=0.1,IPEA=0.3,mix=2,xms=1,maxit=100,maxiti=400
closed,140
occ,152
core,140
wf,287,1,7
state,2}