Coding SNP data for diploid loci
86 views
Skip to first unread message

Sean Canfield
Oct 30, 2021, 2:22:58 AM10/30/21
to migrate-support
Aloha Peter et al.,
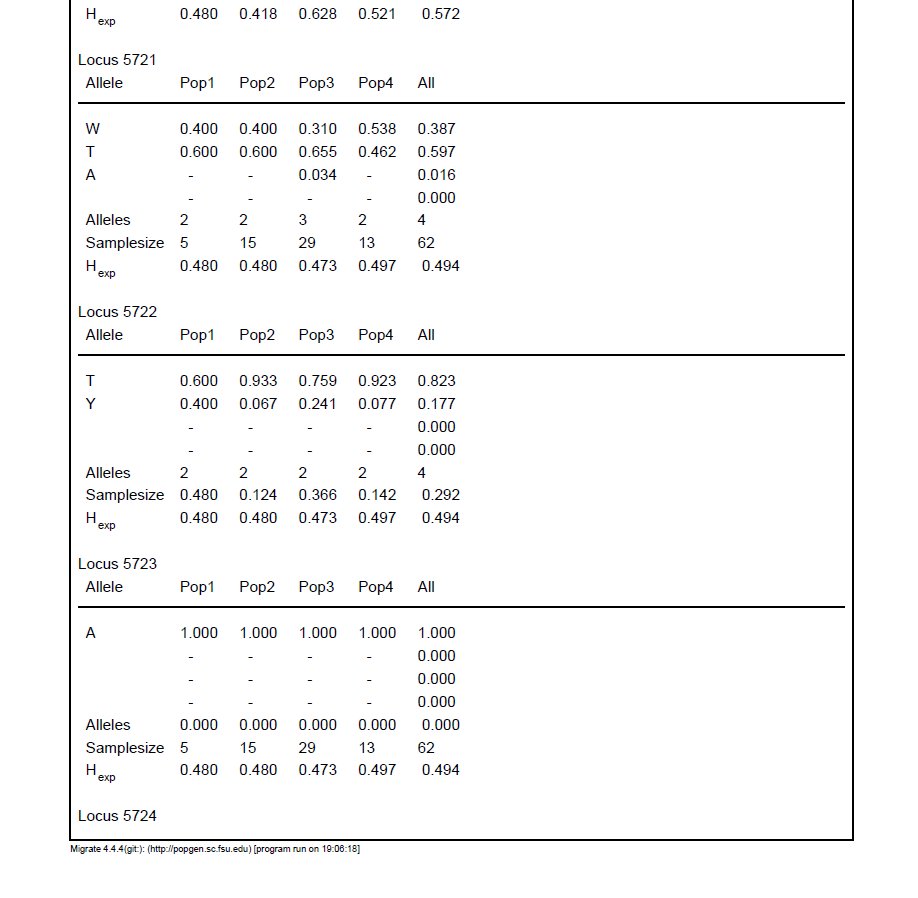
I've converted a VCF into the old MIGRATE format using PGDSpider. Everything looks fine at a glance, and the input file will run. However, when I check the output, MIGRATE treats ambiguity codes as a distinct, third allele. So for example, it will list W, A, and T as three alleles for a single locus, and the sum of the frequencies is 1. For reference, I've included a screenshot of the (massive) output file. Note Locus 5721 near the top of the image (we've already resolved the issue of non-variable loci, such as locus 5723).
Is this a cause for concern? It seems like ambiguity codes are meant to denote a heterozygous genotype in this case (which makes sense), but the allele frequencies are really throwing me for a loop.
Many thanks,
Sean

{kind=link}
Peter Beerli
Oct 31, 2021, 2:38:30 PM10/31/21
to migrate...@googlegroups.com
Dear Sean,
Ambiguity codes in migrate date back to sequence data sets where the IUPAC ambiguity code is used to describe uncertainty.
The ambiguity code in migrate is not used to mark a heterozygote it is used to inform the tree likelihood that we are not sure what
particular nucleotide it is. Therefore the use of ambiguity codes for snps is not useful because it simply says that we do not know which of the two
alleles should be used, your VCF with genotype data actually should emit both alleles and not just one, that could be used to generate two lines in the migrate infile so that in the VCF two individuals with AA, AC would translate in migrate to
one:1 A
one:2 A
two:1 A
two:2 C
two:1 A
two:2 C
The frequency spectra output is inconsequential for the analysis if it prints
W
A
T
that does only affect the frequency tables, but not the run and the Bayes posterior tables.
In the run the likelihood will be calculated from the conditiional tip likelihoods that are set up
using W, A, and T, so your heterzygote is expressed as uncertainty.
If you use large numbers of snp loci
set the parmfile option (you need to edit the parmfile, te menu does not include this) pdf-terse to
pdf-terse=YES
this may help to remedy issues with too large PDF files (that then crash).
Peter
P.S. Eventually migrate will be able to handle VCF data directly, but it is still a long way to go.
--
You received this message because you are subscribed to the Google Groups "migrate-support" group.
To unsubscribe from this group and stop receiving emails from it, send an email to migrate-suppo...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/migrate-support/ee05560d-20b1-40cd-9803-243efd572d4an%40googlegroups.com.
<Output(2)_p2512.jpg>
Sean Canfield
Nov 1, 2021, 4:26:51 PM11/1/21
to migrate-support
Aloha Peter,
Thanks for the quick reply! So will MIGRATE recognize that those two lines represent a single locus? Or will it split each locus into two loci based on this configuration?
- Sean
Sean Canfield
Nov 1, 2021, 4:32:43 PM11/1/21
to migrate-support
Apologies, I should have found this thread earlier:
I think it addresses a lot of my questions. Of course now there's the issue of whether SNP data are ideal for this kind of analysis in the first place, since invariant sites are missing (without full sequences). That's something we'll need to think on a bit more.
Peter Beerli
Nov 1, 2021, 6:21:45 PM11/1/21
to migrate...@googlegroups.com
Sean
I show here a small complete dataset, you see the number of individuals (alleles or gene copies) decides that. In the first population the 2 will be used for each of the 3 loci. If you have different numbers for each locus then you need the second example.
In version 4.x you can use the third example, but that will not allow to
have different number of individuals. The coalescent usually does not link the two alleles of a diploid individual because over long-time they will be in different individuals and are considered independent of each other. I have an experimental haplotyping option for sequences that is rather tricky to set up and therefore is not yet well documented.
Hope this helps
1. example
2 3 oldversion, all loci have 2 individuals
1 1 1
2 pop1
a A
b A
a A
b W
a A
b A
3 pop2
c A
d A
e T
c A
d A
e T
c R
d A
e R
2. example
2 3 oldversion, loci have 2,1,2 and 3,2,1 individuals
1 1 1
2 1 2 pop1
a A
b A
a A
a A
b A
3 2 1 pop2
c A
d A
e T
d A
e T
c R
3. example
2 3 test snp data
(n1)(n1)(n1)
2 pop1
a AAA
b AWA
3 pop2
c AAR
d AAA
e TTR
Peter
To view this discussion on the web visit https://groups.google.com/d/msgid/migrate-support/5821ee3f-bd5b-43a6-a349-a9c05ab57efbn%40googlegroups.com.
Sean Canfield
Nov 27, 2021, 4:37:24 PM11/27/21
to migrate-support
Got it, thanks Peter!
Reply all
Reply to author
Forward
0 new messages