Aligning and calculating pair-wise RMSD using a dynamic selection
160 views
Skip to first unread message

Akshay
Dec 27, 2021, 5:55:27 AM12/27/21
to MDnalysis discussion
Dear Community,
I'm a neophyte to MDAnalysis. It's a really helpful tool for my research. Thanks !
I am trying to calculate the pair-wise RMSD of an interface between two proteins. I have used the following commands as given in the tutorials:
u = mda.Universe("random_frame.pdb","md_corrected.xtc")
interface = '((around 7.5 chainID B) and backbone) or ((around 7.5 chainID A) and backbone)'
aligner = align.AlignTraj(u, u, select=interface,in_memory=True).run()
matrix = diffusionmap.DistanceMatrix(u, select=interface).run()
interface = '((around 7.5 chainID B) and backbone) or ((around 7.5 chainID A) and backbone)'
aligner = align.AlignTraj(u, u, select=interface,in_memory=True).run()
matrix = diffusionmap.DistanceMatrix(u, select=interface).run()
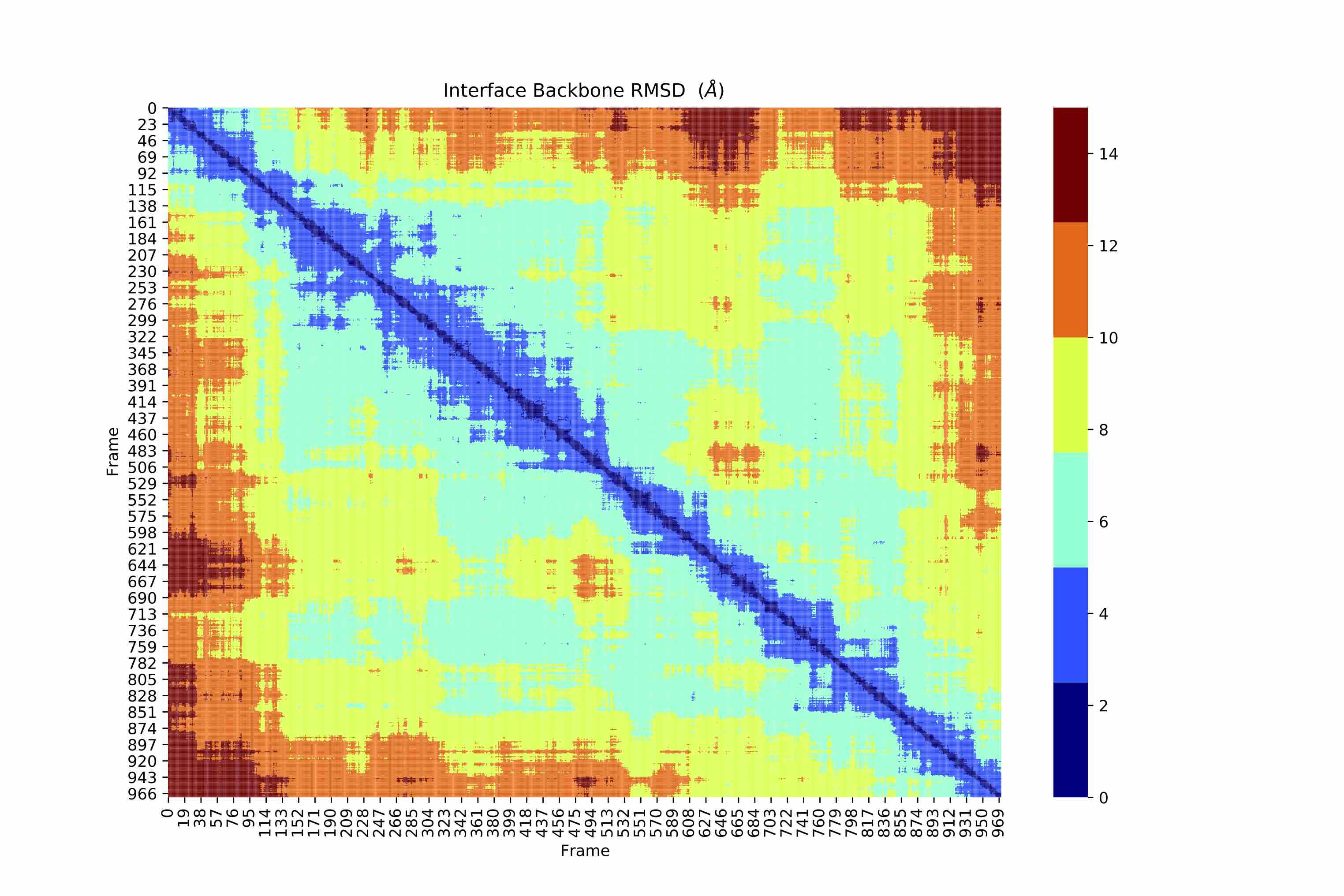
Since the selection is dynamic, I don't expect to see a symmetric RMSD plot (the number of atoms will be different in each frame for a dynamic selection). However the output RMSD graph is symmteric. I am guessing there is something to do with alignment that I don't understand. How does MDAnalysis calculate RMSD when the number of atoms are different in the each frame? Please correct me if I am wrong.
Your insight will be valuable.
Thanks again,

{kind=link}
Lily Wang
Dec 27, 2021, 8:05:36 AM12/27/21
to mdnalysis-...@googlegroups.com
Dear Akshay,
Welcome to MDAnalysis!
Unfortunately dynamic selections are not supported by AlignTraj or DistanceMatrix in that way; when you pass a selection string, only a static selection is made. Therefore, the number of atoms is not different in each frame.This is because both AlignTraj and DistanceMatrix use an atom-wise metric (RMSD), where atom i in group1 is compared to atom i in group 2. So for AlignTraj, the number of atoms used to align the trajectory in each frame must be the same as the number in the static reference. When calculating the RMSD of frame 1 vs. frame 2 in distance matrix, the number of atoms must again be the same.
In your code, you are only choosing the atom group once in the first frame — the backbone atoms near chain A or B. Then, without updating, the trajectory is aligned to the first frame using those atoms, and the RMSD is calculated between each pair of frames.
Perhaps you could find out all backbone atoms near either chain in any frame of the trajectory, and run the analysis on that instead?
ag = u.select_atoms(interface)
nearby_at_any_point = u.atoms[[]]
for _ in u.trajectory:
nearby_at_any_point |= ag
aligner = align.AlignTraj(nearby_at_any_point, nearby_at_any_point, in_memory=True).run()
select_by_index = f"index {' '.join(list(map(str, nearby_at_any_point.ix)))}"
matrix = diffusionmap.DistanceMatrix(u, select=select_by_index)
Cheers,
Lily
--
You received this message because you are subscribed to the Google Groups "MDnalysis discussion" group.
To unsubscribe from this group and stop receiving emails from it, send an email to mdnalysis-discus...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/mdnalysis-discussion/98574272-c7c3-40ae-9554-6baeac7cc264n%40googlegroups.com.
<map_1.jpg>
Akshay
Dec 27, 2021, 9:54:45 PM12/27/21
to MDnalysis discussion
Dear Lily,
Thanks for the detailed reply and for clarifying how align.AlignTraj works.Thanks for the code, I will implement the same.
Immensely appreciate your time and effort in responding to my query.
Lily Wang
Dec 28, 2021, 11:40:57 AM12/28/21
to mdnalysis-...@googlegroups.com
No worries Akshay — I did just notice some typos in my suggested code, apologies for that.
To create a dynamic atom group, you need to pass updating=True:
ag = u.select_atoms(interface, updating=True)
Or, to make it more direct, you could re-create the atom group on each frame:
nearby_at_any_point = u.atoms[[]]for _ in u.trajectory:
nearby_at_any_point |= u.select_atoms(interface)
And I forgot to run() the matrix:
matrix = diffusionmap.DistanceMatrix(u, select=select_by_index).run()
Hope that helps.
Cheers,
Lily
To view this discussion on the web visit https://groups.google.com/d/msgid/mdnalysis-discussion/c104d392-89d9-41e1-8ffb-758cdb293484n%40googlegroups.com.
Akshay
Dec 28, 2021, 10:24:28 PM12/28/21
to MDnalysis discussion
Thanks for getting back Lily, I had noticed these typos and had rectified them in my code.
Reply all
Reply to author
Forward
0 new messages