star 2 pass v. star single pass
1,065 views
Skip to first unread message

arashdeep singh Sran
Mar 21, 2022, 12:49:48 PM3/21/22
to majiq_voila
hi,
I have chosen to generate a bam file using STAR aligner. However, I am confused between two-pass vs. single pass alignment options with STAR.
two pass alignment with STAR would uses the exon junctions generated in the first pass to index the reference genome. Also the MAJIQ manual says that it attempts to learn the exon junctions from the bam file. So could you please clarify which option (2-pass. vs. single pass) is better suited to generate a correct bam file for MAJIQ tool.
Please take a look to help this out.
Thank you and best regards.

Paul Jewell
Mar 23, 2022, 6:28:04 PM3/23/22
to majiq_voila
Hello,
This is an interesting question. I'll attempt to clarify some based on my reading of the STAR manual, however I'm also going to pass this question to some of my peers for possible clarification.
In general, for most of the lab work that we have done, Single-Pass mapping is performed, so for the experience closest to that we are expecting, I would recommend that you use single-pass in your runs.
However conceptually, whether we use STAR in single pass or two pass mode, it will provide us with aligned reads, split over certain positions. ex: found one read which matches half to the location of exon1, and the other half to some intronic location. Or maybe the read is split over three or more locations, etc. The idea being, the end result of STAR is still not any kind of quantified result, but just aligned read-pieces.
This still holds true in two-pass mode. However, by using a lower stringency for the second pass, and also allowing some of the de-novo exons detected by the first pass to be considered "annotated", there will be fewer unmapped read positions, and thus there is potential for more odd splicing patterns to be captured. See this related figure from here, which I believe details a similar approach to STAR: https://academic.oup.com/bioinformatics/article/32/1/43/1744001
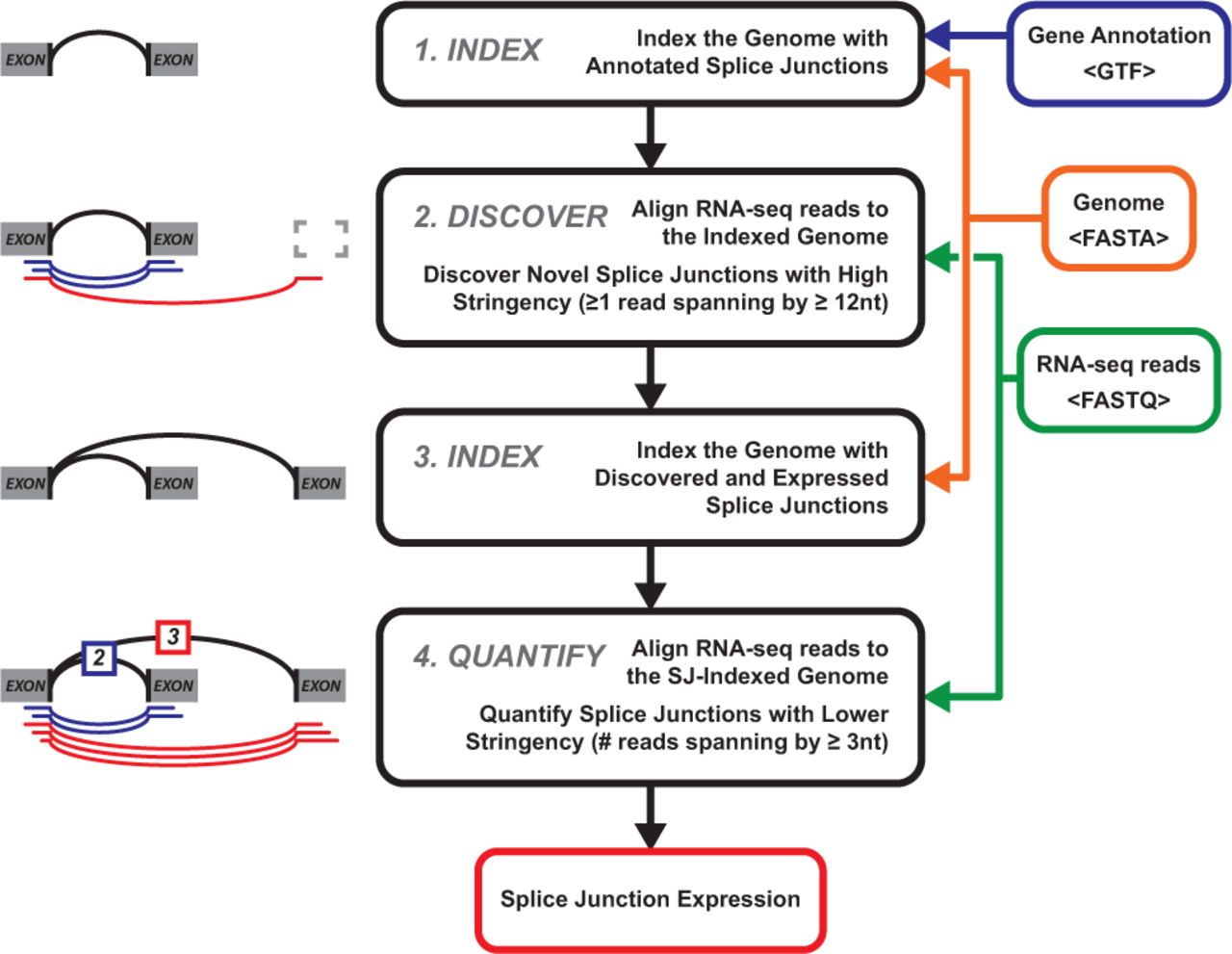
Having more unique mapped reads will help majiq's accuracy when some of these novel alignments may be thrown out by the single pass version.
Paul Jewell
Mar 24, 2022, 9:35:00 AM3/24/22
to majiq_voila
I'll forward another update:
Different members of our lab have different preferences, so I think it's fairly safe to say the big picture will not be dramatically affected by choice of single or two pass alignment. Here are some additional points in favor of one of the two pass modes by one of our heavy Majiq users and contributors Joseph Aicher:
My understanding is that there are multiple versions of STAR two-pass.
The most common (and what I use) is two-pass individual, which updates
the sj database with novel junctions from first pass of the same
experiment alone. Less common alternative is to first pass over group of
experiments, then update sj database with junctions found over all
experiments.
I don't see any reason not to do two-pass individual:
- theoretically, this should help with systematic bias (down) in coverage for denovo junctions
- from a user perspective, all we have to do is add an additional flag to STAR
- the runtime difference is negligible compared to other things we do that probably have similar level of impact (i.e., trimming)
I don't see any reason not to do two-pass individual:
- theoretically, this should help with systematic bias (down) in coverage for denovo junctions
- from a user perspective, all we have to do is add an additional flag to STAR
- the runtime difference is negligible compared to other things we do that probably have similar level of impact (i.e., trimming)
Reply all
Reply to author
Forward
0 new messages