Missing major exon skipping event
39 views
Skip to first unread message

Chris Seward
Sep 1, 2021, 6:28:57 PM9/1/21
to majiq_voila
I've been experimenting with Majiq Build v2.2-e25c4ac to determine if I understand how to use it.
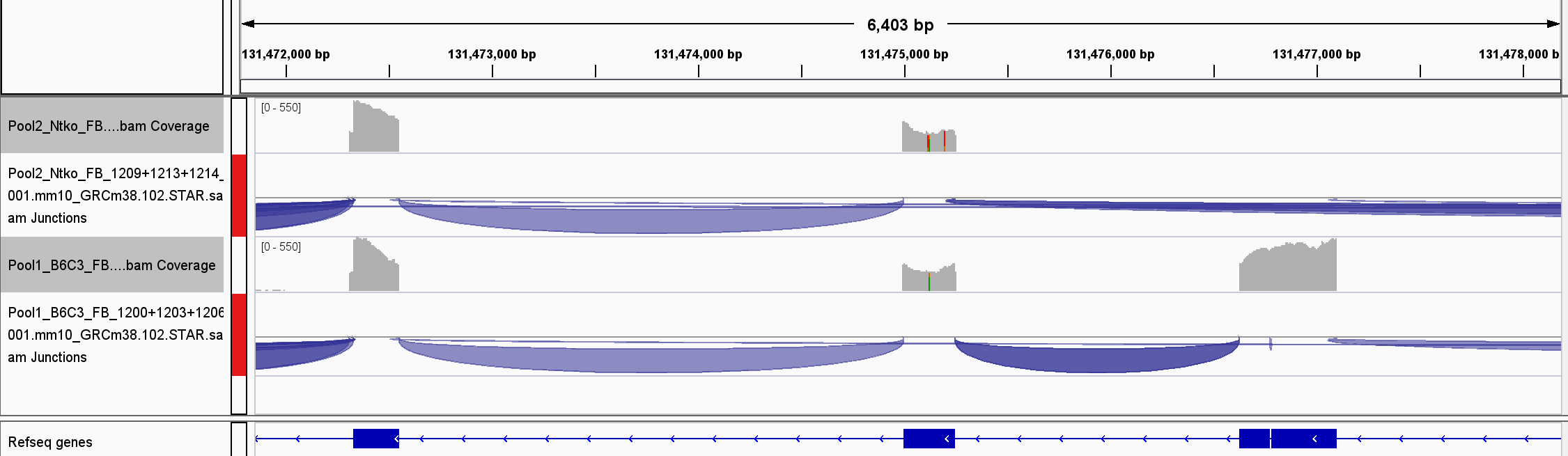
I have a test dataset from two mouse strains, one of which has a knockout of two exons in a gene. We have 9 RNASeq samples for each genotype, with a sequencing depth of 40-50m, paired end, 150bp reads per sample, mapped to the mm10 ensemble genome and ensemble v102 annotation with STAR.
MAJIQ does find a few (~10-20) very minor (hard to spot visually) LSV between the two strains, but they are only located in non-standard chromosomes like chrX_random, chrUn_JH584304 etc. Does this indicate something is off? What is a normal range of numbers of LSV you would expect to find in an actual animal experiment? 100s? 1000s?
I am wondering if MAJIQ is ignoring the actual standard chromosomes for some reason? In particular, it never identifies the actual exon knockout we engineered as a variant.
I can see the variant very clearly myself in the bam files, but it just isn't being spotted computationally. Do you think I am doing something wrong or is this something the program normally wouldn't detect for some reason?
Chris Seward
Sep 1, 2021, 9:13:52 PM9/1/21
to majiq_voila
After more troubleshooting I've determined it was an issue with the genome annotation I was using. I think it was a "chr1" vs just "1" problem (ucsc vs ensemble), but now I've got it working and am finding the top hits I expected to find! It was only working before on the non-standard chromosomes because the nomenclature for those chromosomes is the same between the annotations. Posting this here in case other people have similar issues!

Paul Jewell
Sep 10, 2021, 10:43:34 AM9/10/21
to majiq_voila
Hello,
I'm glad you were able to work out part of the issue. I wanted to mention that there was also a separate longstanding issue people were having with specifying specific strandness in the majiq config file, finding many fewer LSVs than intended. We had a temporary fix for this released in a development branch, but I have just merged it into the master branch today. You can try to reinstall if you were using the strandness specification other than "none" in your config file.
Thanks.
Reply all
Reply to author
Forward
0 new messages