ipi-siesta-fixatoms
236 views
Skip to first unread message

peiwei you
Mar 27, 2020, 2:22:54 AM3/27/20
to ipi-users
Dear all,
While using ipi-siesta, I try to set some atoms fixed and others are not fixed. I checked the temperature and positions at each step and the performed result shows all atoms do not move at all.
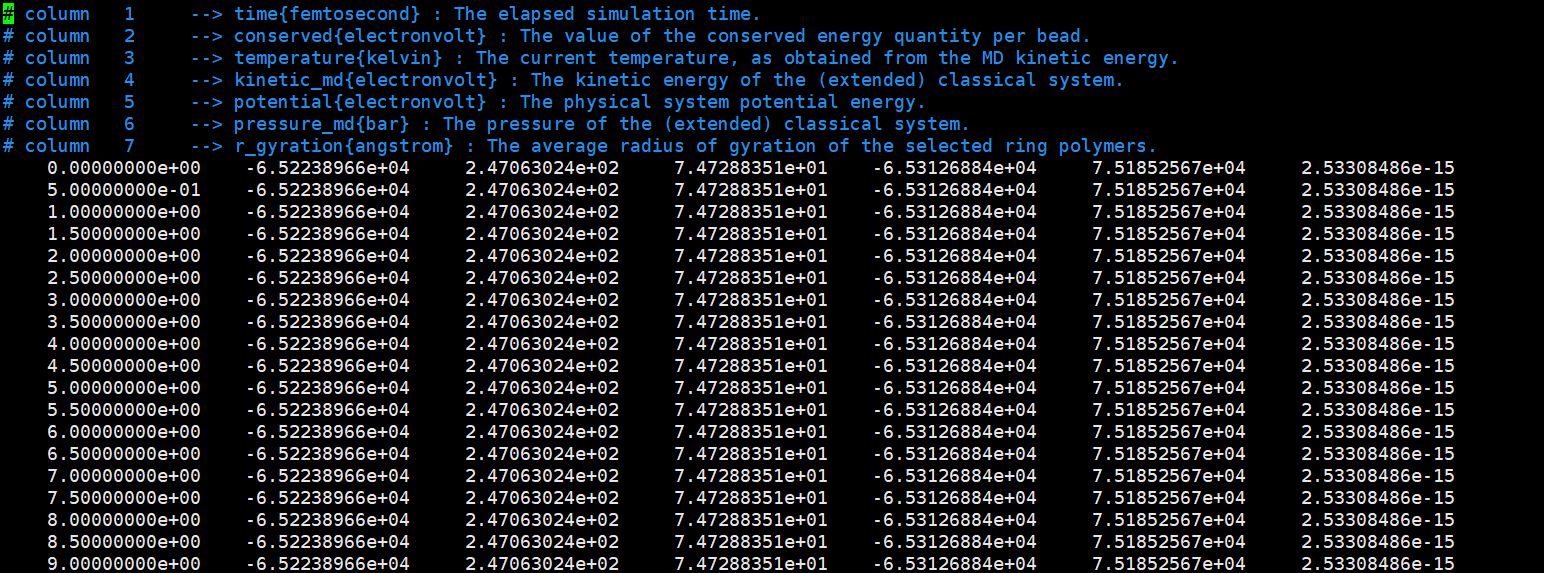
If I delete the <fixatoms> tag, the simulation can be performed as usual.
Does my usage is right or it is a bug of ipi?
Best wishes,
Peiwei You.

Michele Ceriotti
Mar 27, 2020, 3:59:14 AM3/27/20
to ipi-users
This definitely needs further investigation. May I ask you what system is this, how many atoms you have in total, etc?
Might be good to file a bug on github, so we can make sure of what branch you're running on, etc.

Mariana Rossi
Mar 27, 2020, 11:06:54 AM3/27/20
to ipi-users
Dear Peiwei,
Could you share all your inputs/outputs or those of a smaller model system that shows the same problem?
I have previously used the SIESTA interface with fixed atoms and everything behaved normally.
I have previously used the SIESTA interface with fixed atoms and everything behaved normally.
All the best,
Mariana
Mariana Rossi
Mar 31, 2020, 2:35:24 AM3/31/20
to ipi-users
Hi Peiwei,
And the Siesta version you are using is 4.something? Or which version?
Cheers,
Mariana
Mariana Rossi
Mar 31, 2020, 2:42:01 AM3/31/20
to ipi-users
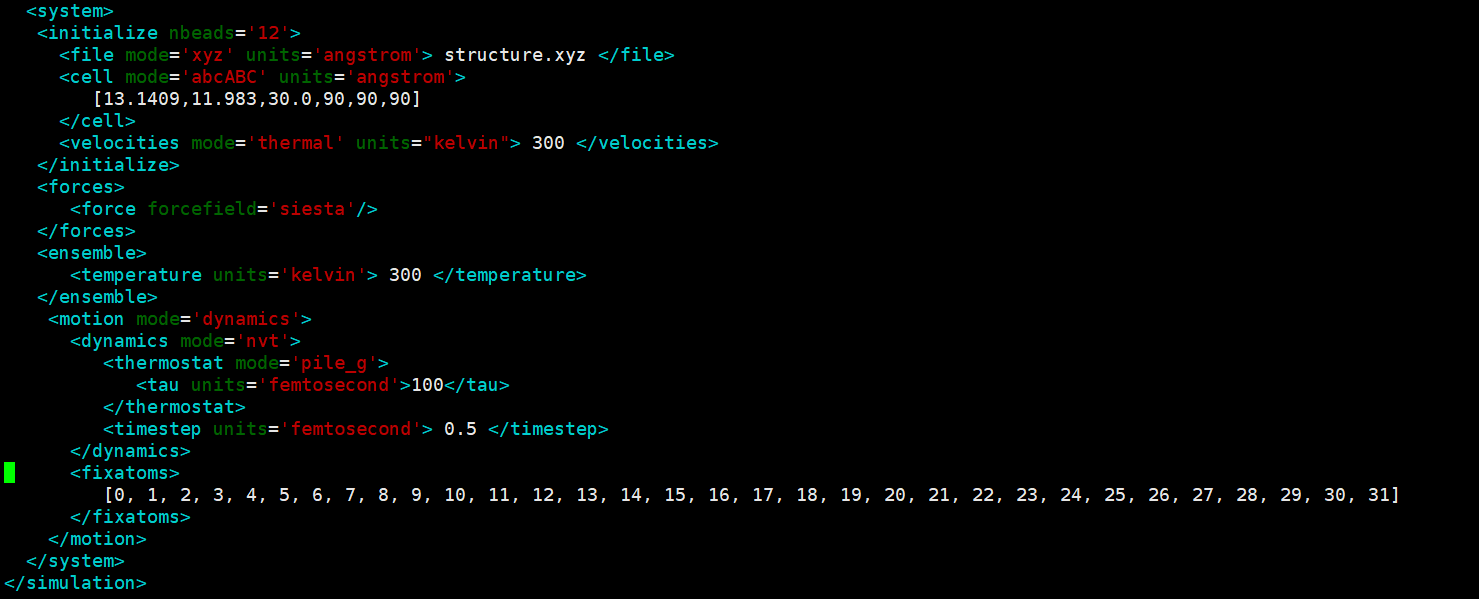
P.S.: Beyond telling me the version of Siesta, could you also please comment out the following blocks and lines in input.fdf and let me know what happens (i-PI takes care of that and this could lead to all sorts of problems):
%block GeometryConstraints
position from 1 to 32
position from 65 to 120
%endblock GeometryConstraints
MD.FinalTimeStep 4000
MD.LengthTimeStep 0.5 fs
MD.InitialTemperature 300 k
Especially the first block will zero forces on atoms that in principle you seem to want to move, if I understood correctly.
peiwei you
Mar 31, 2020, 10:11:44 AM3/31/20
to ipi-users
Dear Mariana,
I'm using siesta-4.1-b4.
The first block
("%block GeometryConstraints
position from 1 to 32
position from 65 to 120
%endblock GeometryConstraints")
means the atom indexs one wanna fix. To test this input block, I've tried to comment them out but it does not work.
MD.FinalTimeStep 4000 # total steps of molecular dynamics.
MD.LengthTimeStep 0.5 fs # time steps of molecular dynamics.
MD.InitialTemperature 300 k # initial temperature of molecular dynamics.
These three inputs doesn't plaly any role while we use ipi as client, since siesta is regared as a force server.
Best wishes,
Peiwei You.
Mariana Rossi
Mar 31, 2020, 12:06:10 PM3/31/20
to ipi-users
Hi Peiwei,
Just to be a bit clearer, the bug is not in the i-PI side. The fixatoms flag works in all situations we test daily, in the regression tests, in relaxations and so on. I am using it with the FHI-aims code today and all is good. Therefore, if there is a bug it is on the SIESTA side, in the way it is taking in the positions it receives from i-PI. I am not a SIESTA developer but I could find some time to check it later in the week just out of curiosity. The simulations I ran were with an earlier Siesta 4 version, almost 1 year ago. I think you could post this in the Siesta forum as well -- but I have not yet tested whether I can reproduce the behavior you see.
The atoms you were constraining in Siesta (65 to 120) were not being equally constrained in i-PI, that is why the blocks were not consistent. Thanks for checking that you do not get it working with that either.
Cheers,
Mariana
Mariana Rossi
Mar 31, 2020, 4:03:54 PM3/31/20
to ipi-...@googlegroups.com
Hi Peiwei,
Taking back what I said above, I ran your system with Siesta and i-PI in my computer and actually got a more informative crash than just your atoms that do not move -- the number of degrees of freedom was going to zero (or becoming negative) in a piece of the code.
After that @ceriotti also had a look at it and found that the problem seems indeed to be with the combination of fixatoms and the pile_g thermostat (!) -- a combination none of us use often at all. We will come back to you tomorrow with more info on this.
Thanks again,
Mariana
peiwei you
Apr 1, 2020, 10:32:59 AM4/1/20
to ipi-users
Dear Mariana,
That is really helpful! Thanks very much. I've tested another thermostat (langevin) and it seems <fixatoms> tag works well. The bottom two layers are fixed and other atoms are moving.
By the way, what thermostats do you often use?
Best wishes,
Peiwei You.
Mariana Rossi
Apr 1, 2020, 11:53:46 AM4/1/20
to ipi-users
Dear Peiwei,
This PR https://github.com/i-pi/i-pi/pull/85 should fix your troubles and you could continue using pile_g. Could you please check that it does? It works for me now, but you had a different error. You should be able to clone that branch. If not, let me know.
Note that you will have to add to your i-PI input file the line <fixcom> False </fixcom>. The reason is that now i-PI will stop if you try to have fixed center of mass and fixed atoms (which break translational symmetry) at the same time. We may
improve the handling of this in the near future, but for now please add that line _if_ you want to fix atoms in your simulation.
In your simulation, if you are free to use any thermostat, pile_l for example is perhaps a better choice (local thermostat on the centroid) or any sort of nm_gle. However, it is not a must, and things should be fine also with pile_g now.
Cheers,
Mariana
Mariana Rossi
Apr 3, 2020, 10:13:41 AM4/3/20
to ipi-users
Dear Peiwei,
After revising the PR mentioned above. we concluded that fixing this properly is rather convoluted given the architecture of i-PI.
The previous fix would work for your case but not when just fixing the center of mass of the system. Therefore we decided to
disable the functionality (global thermostat on the centroid _and_ fixed atoms simultaneously) n the py2 master branch and work on a deeper fix involving some rewrite of the architecture in the py3 version.
We suggest you use the thermostat pile_l for your simulations. Please let us know if you need any further help setting up.
All the best,
Mariana
peiwei you
Apr 4, 2020, 12:01:17 PM4/4/20
to ipi-users
Dear Mariana,
I've tested <fixatoms> and <fixcom> False </fixcom> tag while using pile_g thermostat. It works! The bottom two layers are fixed and other atoms are moving.
Thanks a lot for your help.
Best wishes,
Peiwei You.

Fernand Louisnard
Feb 1, 2023, 12:53:26 PM2/1/23
to ipi-users
Dear all,
having the same bug in my simulation, I would like to know if it is okay to use the pile_l thermostat for TRPMD simulations with fix atoms in order to compute IR spectra.
Best,
Fernand Louisnard
marian...@gmail.com
Feb 2, 2023, 4:36:09 AM2/2/23
to ipi-users
Hi Fernand,
Yes, choose a very large tau for pile_l and it should be equivalent.
All the best,
Mariana
Reply all
Reply to author
Forward
0 new messages