ChIP-Seq pipeline in "tf" mode
100 views
Skip to first unread message

Asma Riyaz
Oct 14, 2020, 9:36:14 AM10/14/20
to idr-discuss
Hello,
I have a couple of questions regrading the output and its interpretation:
1) The peak files obtained are suffixed with "regionPeak" and not "narrowPeak". Could you help me understand why this would be the case? The output of call-reproducibility_idr has 10 columns and not 9 (as is mentioned in the format description for broad/region peak). Here is an excerpt from the output.
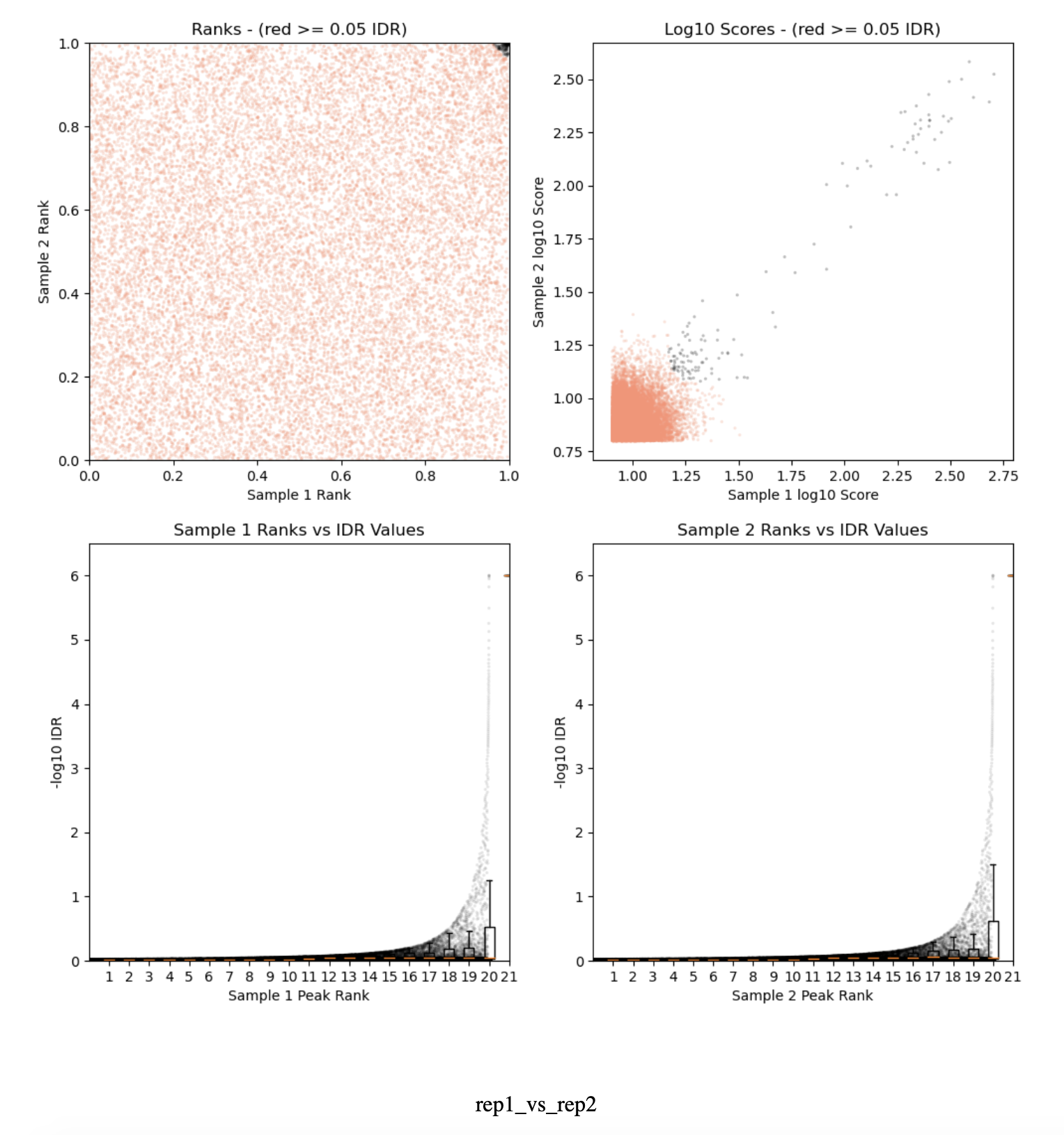
2) The IDR plots of all 3 samples show very little black dots as compared to the tutorials I have found online. Why would this be the case? Is it because the regions found from this protocol are broad instead of narrow? I have attached a plot here for reference.
3) While I understand the conservative peaks refer to peaks only from the replicates and optimal peaks come from both replicates and pooled samples, I was hoping to understand which set to consider for downstream analysis as there are very few black dots in my case in the IDR plot?
I have attached the qc report for one of the samples BZW1 with this thread in case you may find it useful to help me.
Thank you,
Asma
{kind=link}
Reply all
Reply to author
Forward
0 new messages