Z Score is None in Enrichment Analysis with KEGG pathway
120 views
Skip to first unread message

George
Feb 12, 2016, 4:48:44 PM2/12/16
to Gitools users
Hello,
I try to do an enrichment analysis on my human protein expression
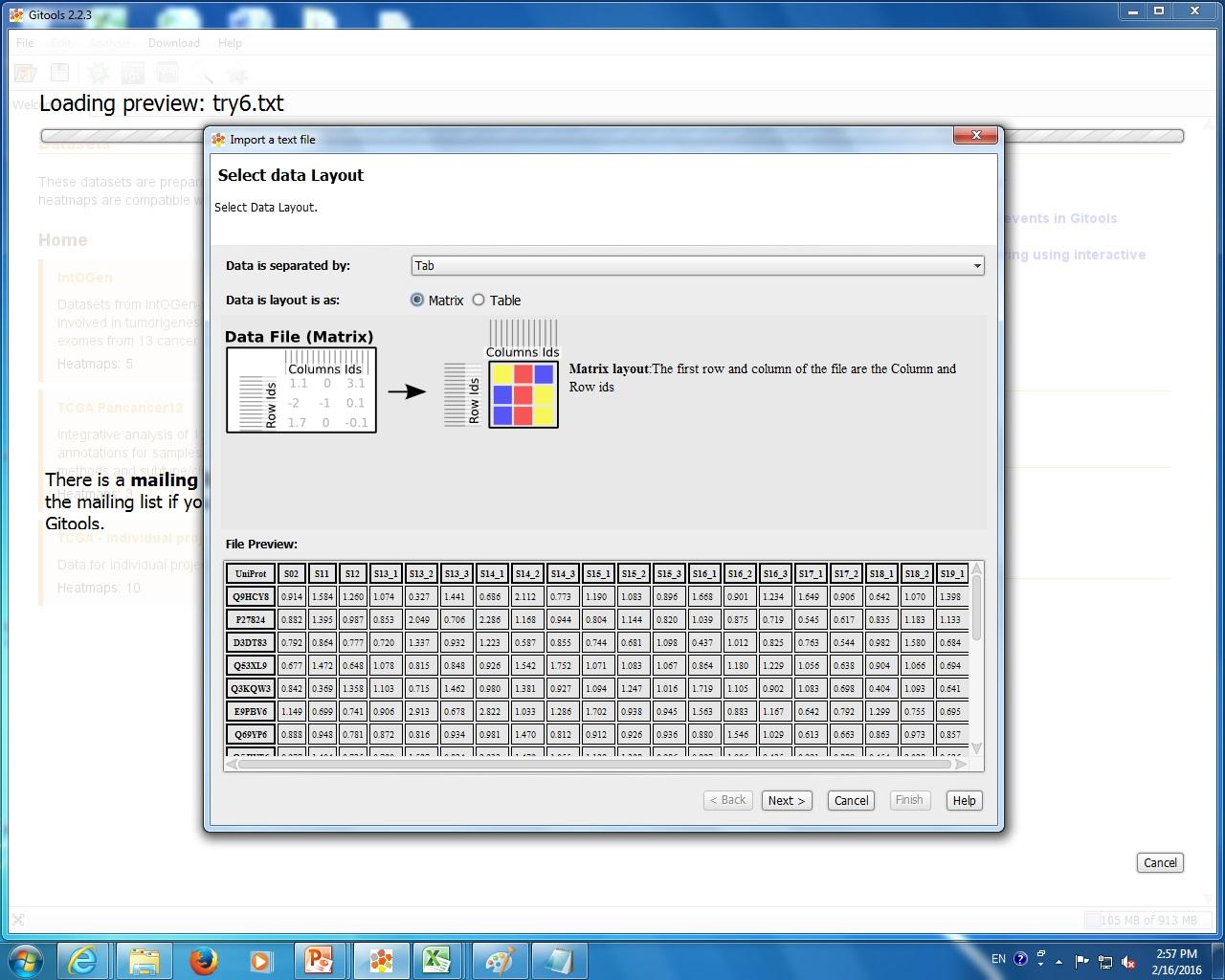
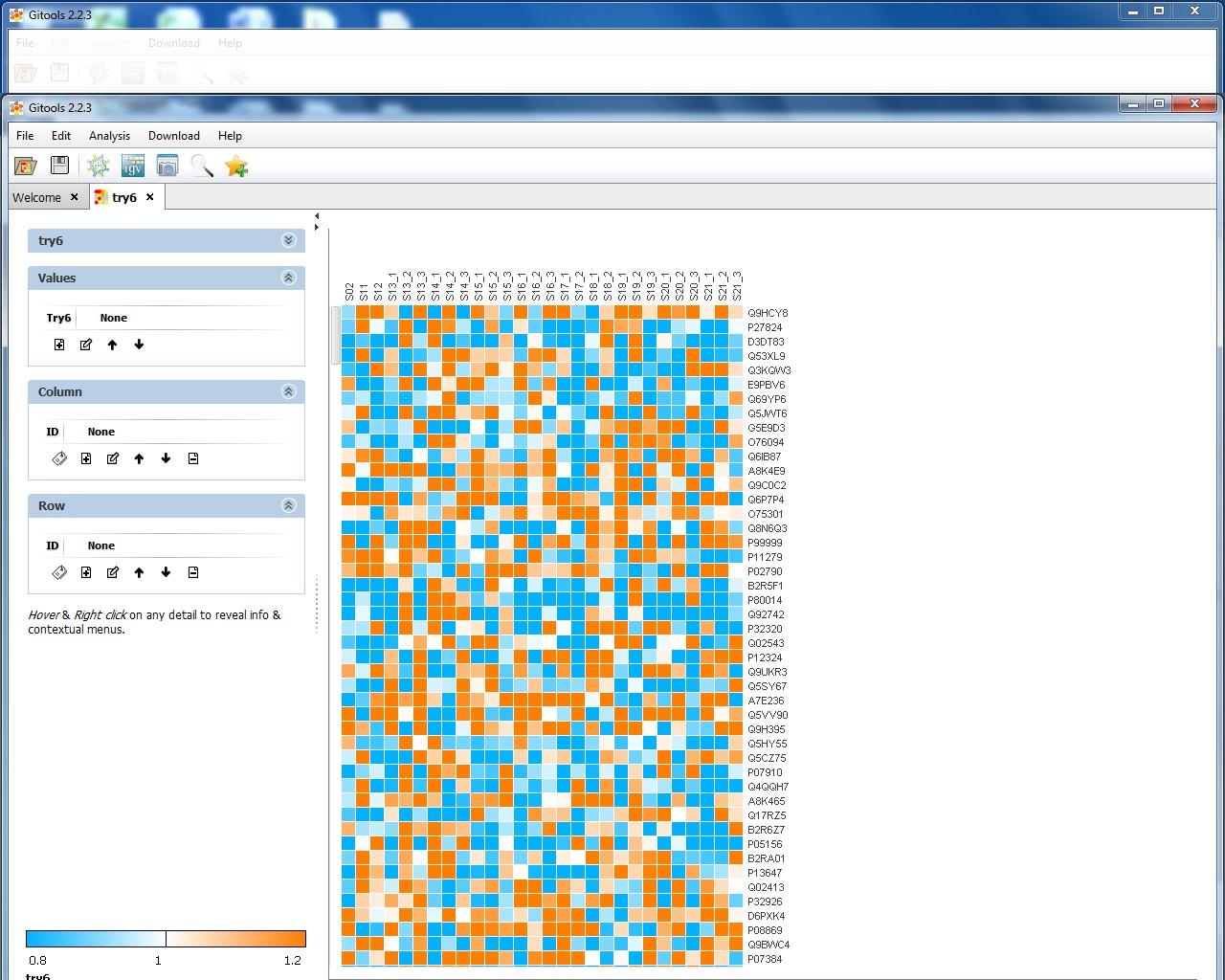
data. I have no problems loading my txt file and I can see the heat map showing the
data for all the different proteins. However, when I do the analysis with the human KEGG
pathway files I downloaded, I can't see anything in the resulting heat
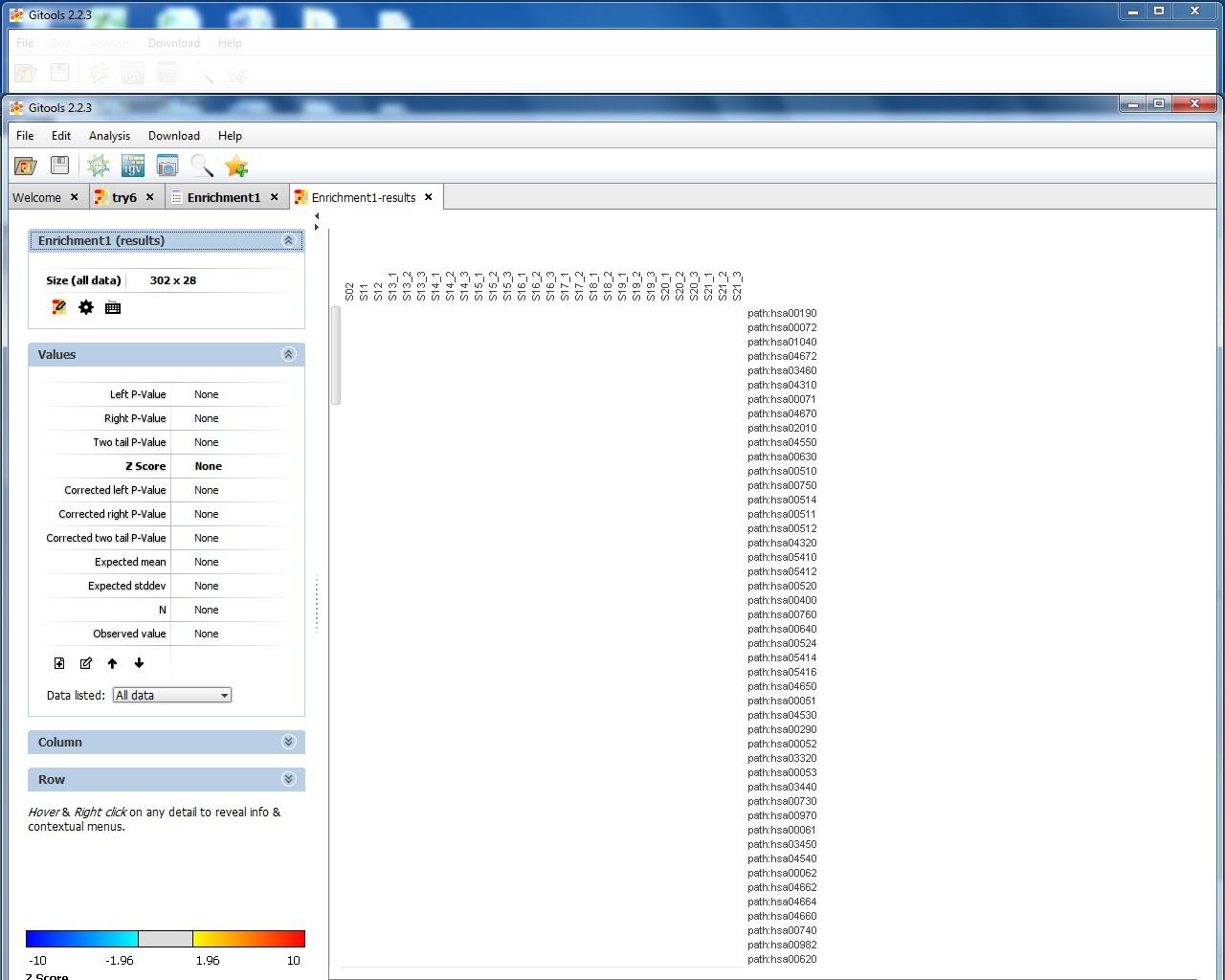
map except sample ID and pathway ID. The module I used should be correct 'cause the protein accession ID is UniProt (for example: Q9HCY8) and I download same module. I notice in the value section everything is none (including p-value and z score). My data is the ratio of protein expression (like 2.5, 0.7, etc). Is there something wrong? Thanks.
Best,
George

Michael P Schroeder
Feb 15, 2016, 3:20:05 AM2/15/16
to Gitools users
Hi George
Could you tell us what cut-offs for module size and so on you chose?
There are one possible problems I can think of from the top of my head: There is not enough data for the modules - meaning that in the case that you require 20 items per Module (default setting) and only very fewer proteins are mapped to the respective pathway.
Other than that, it's hard to know what your problem may be without any further information. Did you check that the Ids in the pathway file match the Ids of the heatmap? What tests and cut-offs did you choose? Maybe you can send a screenshot of the analysis page?
Best,
Michael
--
You received this message because you are subscribed to the Google Groups "Gitools users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to gitools-user...@googlegroups.com.
Visit this group at https://groups.google.com/group/gitools-users.
George
Feb 16, 2016, 3:35:12 PM2/16/16
to Gitools users
Dear Michael,
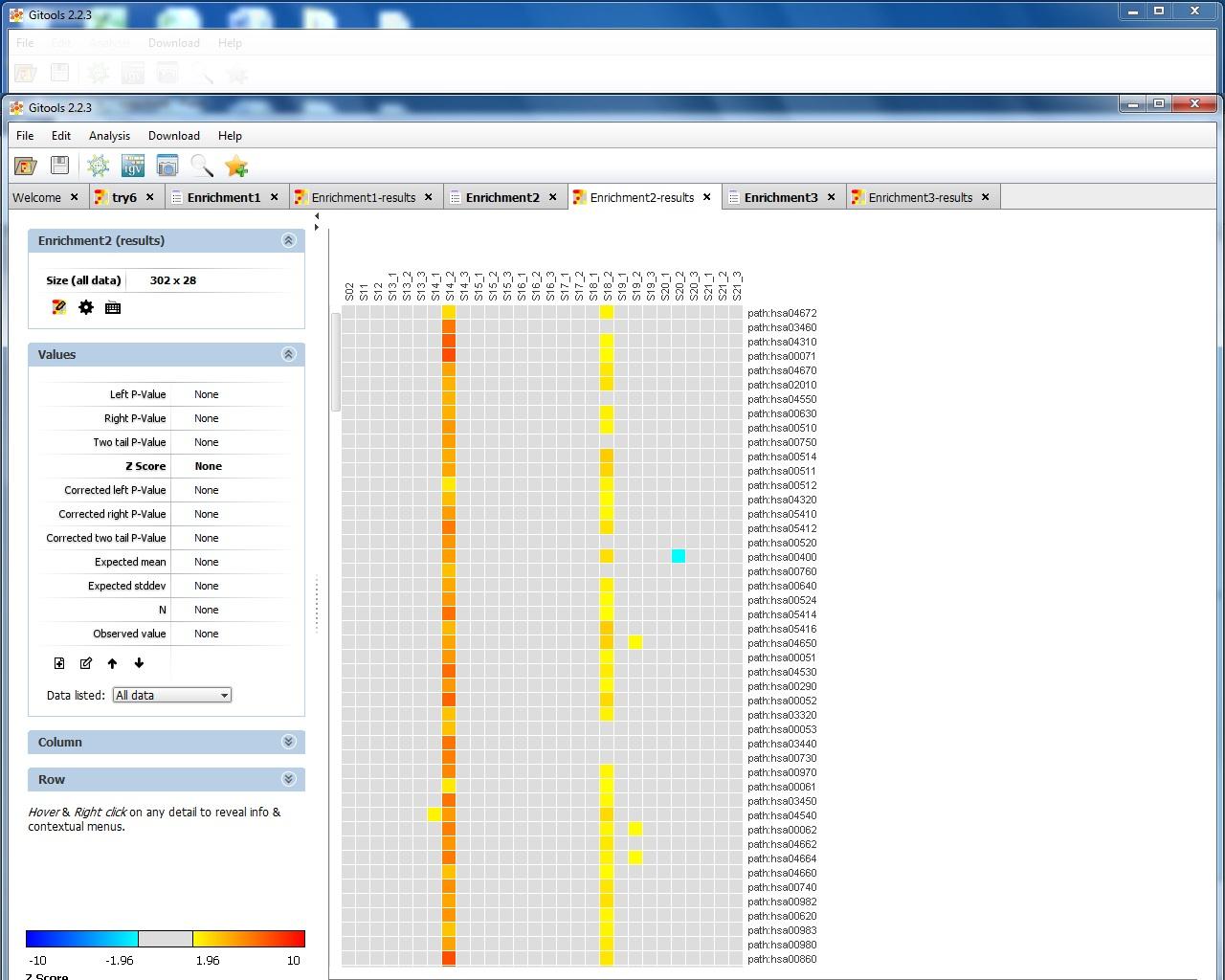
I attach 5 figs show the process of my Gitools analysis. Fig.1 show the file format of my input data. Fig.2 show the heat map of the value of my data. Fig3. show the enrichment result when I use "homo_sapiens__kegg_pathway__uniprot_protein.tcm" module with cut-offs 20. Fig.4 shown the enrichment result when I use "homo_sapiens__kegg_pathway__uniprot_protein.tcm" module and uncheck "omit the modules having annotated rows less than" option. Fig.5 show the enrichment result when I use "homo_sapiens__kegg_pathway__uniprot_protein.tsv" module and uncheck "omit the modules having annotated rows less than" option. I notice that when I select .tsv file as module, the error message "the file extension doesn't match the selected format" appeared, but in the result heat map it show pathway name. However, when I select .tcm file as module, though no error message appear, but in the result heat map, it only show pathway ID like hsa00071. The test I selected is Z-score and sample size is 100.
Could you please figure out the problem base on the screenshot I provide? Please let me know if you need more details. Thanks.
Best,
George
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Michael P Schroeder
Feb 17, 2016, 5:05:42 AM2/17/16
to Gitools users
So the the first problem is that you have less than 20 data points per Module which leads (with the cut-off set) that the test is not performed because it may be difficult to trust.
The identifier that will be shown in the result heatmap depends on the identifiers in the mapping file: If you the file maps Uniport Ids to KEGG ids (e.g. hsa:012212) these Kegg Ids will be shown in the heatmap. Note that there should be an annotation file that has been download with the .tcm file which contains the KEGG pathway names so you can add it as a text label.
You other file seems to map directly to the pathway names, which seems to work fine also. What is weird is that the results differ so much between the two mappings! Could you load the pathway names in the heatmap with only hsa-identifiers and check if the results coincide?
Also to check if the modules are the same between the two annotation, select one or two pathwyas and check if the annotated UNIprot Ids coincide?
Reply all
Reply to author
Forward
0 new messages