Ask for suggestions on dealing with big gene trees

Yiyan Yang

Benoit Morel
Benoit Morel
Yiyan Yang
- parallelization will reduce the runtime a lot, so if you have a big machine or a cluster available, use it (even if there is only one gene tree to estimate).
Benoit Morel
--
You received this message because you are subscribed to the Google Groups "GeneRax" group.
To unsubscribe from this group and stop receiving emails from it, send an email to generaxusers...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/generaxusers/4a87c6b1-2119-4dde-b122-1c4bf6f71c66n%40googlegroups.com.
For more options, visit https://groups.google.com/d/optout.
Yiyan Yang
Dear Benoit,
Thanks a million for your super valuable suggestion!
I would also like to introduce you more about what I am trying to achieve in my project and ask for your opinion on how GeneRax could be involved in it.
I have collected a group of genomes and annotated them with trait data (such as soil, maine etc habitats). A species tree was built with the genomic data (annotating trait with different colors):
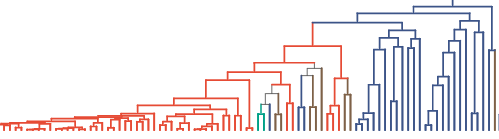
My plan is to infer DTL rates for red and non-red branches so that a group of genes with significantly different DTL rates between red and non-red branches could be found and further analyzed. My final goal is to see how these red genomes/species evolve to adapt to their unique habitat. This can also be extended to other scenarios in Comparative Biology and various hypotheses could be tested. Then I found GeneRax and thought this new and fantastic tool may be very helpful.
That is why I am wondering [Question 1] if there is a single rate estimated for all the red branches (this is where my first question about branch-specific rates came from). After discussing with you, I think the "--per-species-rates" option (i.e. free rate model) is definitely a good choice, yet considering its complexity and my use of big species/gene trees, a “partially free rate” model may greatly reduce the computation time and seems more suitable in this case.
Naturally, I am also wondering [Question 2] if the DTL rates can be compared between different branches. In other words, is it reasonable or meaningful to compare them?
At last, would you mind taking a very quick look at the following command lines I constructed according to your suggestion to make sure I understand you correctly?
STEP1:
mpirun -np $cores $generax --families $families_2 --species-tree $speciestree --rec-model UndatedDL --prefix $output_1 --max-spr-radius 3 --strategy SPR
STEP2:
# Save the output gene trees from STEP1 and change the gene tree paths in $familiesmpirun -np $cores $generax --families $families_2 --species-tree $speciestree --rec-model UndatedDL --per-species-rates --prefix $output_2 --max-spr-radius 3 --strategy SKIP
I may have asked many questions in a single conversation and I truly and always appreciate your patience and help.
Best,
Yiyan
Yiyan Yang
Yiyan Yang
# Save the output gene trees from STEP1 and change the gene tree paths in $families
Benoit Morel
Yiyan Yang
1. generax --families $families --species-tree $speciestree --rec-model UndatedDTL --prefix $output --max-spr-radius 3 --si-strategy SKIP