--reconciliation-samples and trasnfers.txt files
49 views
Skip to first unread message

Neha Sahu
Aug 10, 2022, 6:28:23 AM8/10/22
to GeneRax
Hi Benoit,
I am running generax with the option of --reconciliations-samples-100. I am interested in the transfers.txt file that has the donor and recipient nodes for the inferred transfers.
The output reconciliations folder has the following transfers.txt files
A : 100 transfer.txt files (eg OG00001_0_transfers.txt, OG00001_1_transfers.txt, OG00001_2_transfers.txt ..... OG00001_100_transfers.txt)
B : a simple OG00001_transfers.txt file
I couldn't find a documentation for the --reconciliation-samples parameter and the effect it has on the events.newick file..
Could you please describe how the "B" file is generated in this case .. should it contain all the transfers that happen in "A"....because on checking the files, it seems like B is a subset of A.
Thanks in advance,
Neha

Benoit Morel
Aug 10, 2022, 4:13:41 PM8/10/22
to Neha Sahu, GeneRax
Hi Neha,
This option is not documented because it is experimental.
Here is what it does: for a given gene tree topology, there are many possible reconciliation scenarios. By default, GeneRax looks for the maximum likelihood scenario. With this option, it randomly samples those possible reconciliation scenarios instead of just picking the best one.
But you have to keep in mind that the gene tree topology is fixed, so we account for the reconciliation uncertainty, but not for the tree topology uncertainty.
B is the maximum likelihood scenario. The other files are random samples.
Does it answer your question?
Benoit
--
You received this message because you are subscribed to the Google Groups "GeneRax" group.
To unsubscribe from this group and stop receiving emails from it, send an email to generaxusers...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/generaxusers/a84da185-e310-4ec1-90c2-4166932300f0n%40googlegroups.com.
For more options, visit https://groups.google.com/d/optout.
Neha Sahu
Aug 11, 2022, 4:11:01 AM8/11/22
to Benoit Morel, GeneRax
Hi Benoit,
Thank you for the clarification.
Yes, it does.
So, I guess the events.newick file would remain the same with or without this option..?
Also, do you have any experience on how this option affects the run times.
Regards,
Neha
Neha Sahu
Benoit Morel
Aug 11, 2022, 4:22:16 AM8/11/22
to Neha Sahu, GeneRax
As far as I remember, all the other files should remain the same. In addition, there should be an additional file OG00001_samples.nhx with one reconciled tree per sample in the nhx format.
In my experience, this does not affect the runtime at all, because the reconciliation step is much faster than correcting the gene tree topologies. But let me know if you observe a slow down with this option.
Neha Sahu
Aug 11, 2022, 4:39:32 AM8/11/22
to Benoit Morel, GeneRax
Okay, that's great! Yes, so far I didn't encounter any slow downs.
Thank you so much for your help. :)
Cheers,
Neha
Neha Sahu
Neha Sahu
Sep 16, 2022, 3:53:39 AM9/16/22
to GeneRax
Hi Benoit,
In the reconciliations directory, there's a file called transfer.txt - which contains donor and recipient species for the transfer events. Is there a way to know the gene/protein IDs for these events as well.. as how they are mentioned in the reconciliation.xml files?
Thanks in advance,
Neha
Benoit Morel
Sep 16, 2022, 3:58:09 AM9/16/22
to Neha Sahu, GeneRax
Hi Neha,
So far that's not possible, but maybe I could do something about it. The problem is that the transfers can happen at the internal nodes of the gene tree (and those nodes don't have a gene ID). But I could generate a name for each internal node of the output gene tree and output those names in another transfer file. What do you think?
Best,
Benoit
To view this discussion on the web visit https://groups.google.com/d/msgid/generaxusers/c7d81b01-38c6-4607-92dd-451baaae3c1bn%40googlegroups.com.
Neha Sahu
Sep 16, 2022, 4:44:45 AM9/16/22
to GeneRax
Yes, I agree the internal nodes are tricky.
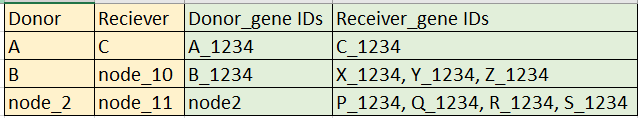
What you suggested sounds like a good idea and would be really helpful, however, I was thinking for 100s of output gene trees, would it be confusing to process each gene tree's internal nodes (because the names would overlap)?
The way I was picturing this is something like:
- what we have now is the yellow region in the transfer.txt - that's based on the species tree node labels - this is very nice and easy to compare across multiple gene trees, and for something like the 1st row, its fairly simple to get the donor and recipient genes
- for 2nd row, when transfer happened from 1 gene of B species to 3 genes in node_10 (listed in Reciever _geneIDs) - this info can be taken from the recphylo.xml
- however third row, where transfer happened between two internal nodes, I guess mentioning just the recipient genes to which transfers were inferred would be cool, and maybe the donor can remain as node2 itself..because the recipient genes IDs are more important here, for listing the exact horizontally transferred genes.
I don't know if it makes sense to do it this way.. (I'm still learning generax :))
Or if there's a way to parse the recphylo.xml files that would be fine too I guess (because those files have the geneIDs and the internal node labels based on species tree - which remains the same throughout)
Regards,
Neha
Benoit Morel
Sep 18, 2022, 4:35:18 PM9/18/22
to Neha Sahu, GeneRax
Hi Neha,
Maybe there is a confusion with what the --reconciliation-samples does: it only produces one gene tree topology (we don't output a distribution of gene trees, but rather the maximum likelihood gene tree). Then, we sample 100 times the reconciliation scenarios that are compatible with the input species tree and this gene tree topology. So there should not be any overlap problem.
The problem with outputting a list of descendant leaves is that it can produce large files for trees with many leaves. That's why I would prefer to use gene IDs, even for internal nodes.
Best,
Benoit
To view this discussion on the web visit https://groups.google.com/d/msgid/generaxusers/2cece148-2a2d-4399-9531-6adbc47981c4n%40googlegroups.com.
Neha Sahu
Sep 19, 2022, 10:19:48 AM9/19/22
to GeneRax
Hi Benoit,
Thank you for the clarification.
Actually, I was talking about overlapping between gene trees of different orthogroups, and not the 100 random reconciliation scenarios...(1 maximum likelihood-based genetree for each orthogroup).. but I guess that could be solved with some data wrangling.
Yes, in any case, I think it would be a nice addition if we could get the list of geneIDs for the transfer events (donor/recipients) along the transfers.txt files.
Would be glad to try if such an update comes up in the newer versions.. :)
Thanks and Regards,
Neha
Reply all
Reply to author
Forward
0 new messages