Comparison of results from exciting and wien2k
161 views
Skip to first unread message

Natalie Holzwarth
Nov 27, 2012, 7:50:32 PM11/27/12
to excit...@googlegroups.com
Dear exciting groups,
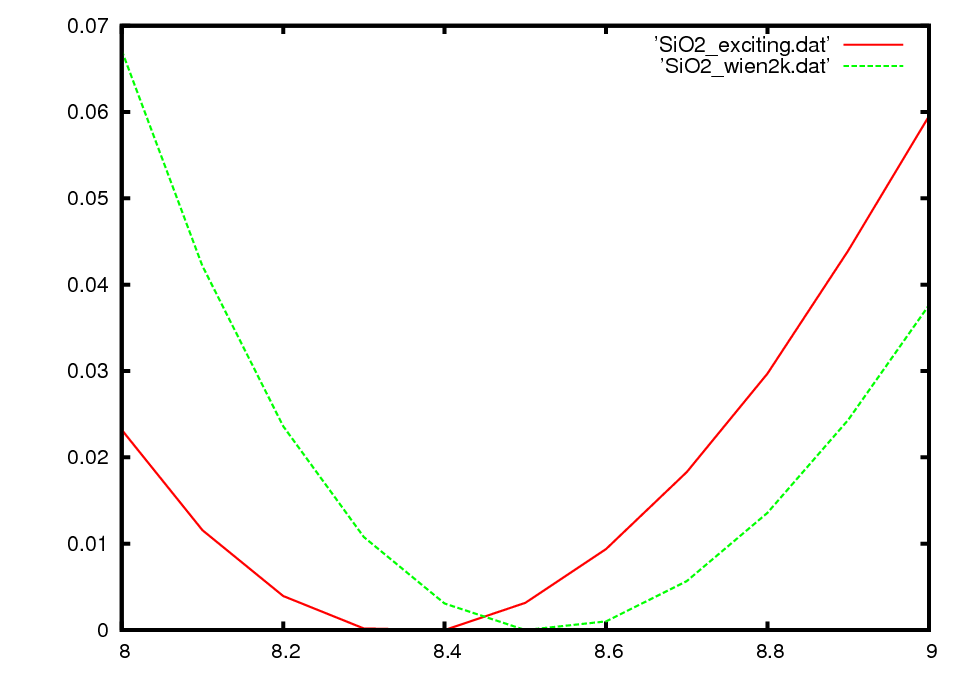
Thanks very much for sharing your code. So far I have found it to be easy to use compared to wien2k. Unfortunately so far, the results using exciting are quite different from what I believe are well-converged results for the same material using wien2k. I am not sure whether you could perhaps spot some silly errors in my use of your code. The test case is a fake fluorite stucture with the composition of SiO2. the input.xml file and a bashscript used to generate the attached plot are given below. I used the species files supplied on line and mostly default parameters. (The rmt parameter did not seem to have an effect.) The attached plot shows the binding energy (Ry) versus lattice constant (in bohr) for exciting and for wien2k. Thanks in advance for any suggestions. SIncerely, Natalie Holzwarth
input.xml:<input>
<title>SiO2 fakefluorite </title>
<structure speciespath="/home/natalie/EL6/publiccode/exciting/excitingNov2012/species/">
<crystal scale="8.0">
<basevect>0.0 0.5 0.5</basevect>
<basevect>0.5 0.0 0.5</basevect>
<basevect>0.5 0.5 0.0</basevect>
</crystal>
<species speciesfile="Si.xml">
rmt="1.5"
<atom coord="0.00 0.00 0.00" />
</species>
<species speciesfile="O.xml">
rmt="1.5"
<atom coord="0.25 0.25 0.25" />
<atom coord="-0.25 -0.25 -0.25" />
</species>
</structure>
<groundstate ngridk="8 8 8"
swidth="0.0005"
rgkmax="8.0"
xctype="LSDAPerdew-Wang">
</groundstate>
</input>
#!/bin/bash
excitingser
cp INFO.OUT INFO.OUT.8.0
echo -n 8.0 > lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.1"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.1
echo -n 8.1 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.2"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.2
echo -n 8.2 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.3"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.3
echo -n 8.3 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.4"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.4
echo -n 8.4 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.5"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.5
echo -n 8.5 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.6"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.6
echo -n 8.6 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.7"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.7
echo -n 8.7 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.8"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.8
echo -n 8.8 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="8.9"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.8.9
echo -n 8.9 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
sed 's/crystal scale="8.0"/crystal scale="9.0"/' input.xml.save >input.xml
excitingser
cp INFO.OUT INFO.OUT.9.0
echo -n 9.0 >> lattice.dat
grep 'total energy ' INFO.OUT |tail -1 >> lattice.dat
output -- lattice.dat
8.0 total energy : -438.717587265
8.1 total energy : -438.723398714
8.2 total energy : -438.727210530
8.3 total energy : -438.729088319
8.4 total energy : -438.729178866
8.5 total energy : -438.727603924
8.6 total energy : -438.724489210
8.7 total energy : -438.720023768
8.8 total energy : -438.714328367
8.9 total energy : -438.707209496
9.0 total energy : -438.699429264

Pablo García Risueño
Nov 28, 2012, 4:55:24 AM11/28/12
to excit...@googlegroups.com
Dear friend
I afraid I cannot properly understand your problem. In the picture you display, both curves look pretty similar. Could you explain us a little bit more about the problem.
You can increase the size of your basis by increasing the parameter rgkmax (e.g. to 9), what should result in greater accuracy.
Best regards
I afraid I cannot properly understand your problem. In the picture you display, both curves look pretty similar. Could you explain us a little bit more about the problem.
You can increase the size of your basis by increasing the parameter rgkmax (e.g. to 9), what should result in greater accuracy.
Best regards
Pablo García Risueño
Nov 28, 2012, 9:02:34 AM11/28/12
to excit...@googlegroups.com
Hello! A mate points what follows. He says that the unit vectors of Exciting are wrong, because at your system the unit cell has a .neq. c:
Two things at first sight Cell Dimensions for Quartz: a = 4.9133, c = 5.4053 (Angstrom)
reference http://www.webmineral.com/data/Quartz.shtml
No idea what kind of W2K has been done
Natalie Holzwarth
Nov 28, 2012, 12:37:37 PM11/28/12
to excit...@googlegroups.com
Sincerely, Natalie Holzwarth
Pablo García Risueño
Nov 28, 2012, 12:55:25 PM11/28/12
to excit...@googlegroups.com
Some other mate recommended to send the input and species files from Exciting and Wien2k, too (so that everybody can check that your input systems are as similar as possible).
Natalie Holzwarth
Nov 28, 2012, 1:44:10 PM11/28/12
to excit...@googlegroups.com
Dear Pablo,
Thanks for your suggestion. For exciting, I used the species file from the download tar file which I believe is the same as the one on line. For wien2k, I have attached some of the input files. A modified optimize script was used to run with the same lattice constants as for exciting. thanks again for your feedback. Natalie Holzwarth Actually -- google would not let me attach the files so I list them below
SiO2.in0
TOT 5 (5...CA-LDA, 13...PBE-GGA, 11...WC-GGA)
NR2V IFFT (R2V)
36 36 36 2.00 1 min IFFT-parameters, enhancement factor, iprint
SiO2.in1
WFFIL EF=.56485 (WFFIL, WFPRI, ENFIL, SUPWF)
9.00 12 6 (R-MT*K-MAX; MAX L IN WF, V-NMT
0.30 3 0 (GLOBAL E-PARAMETER WITH n OTHER CHOICES, global APW/LAPW)
1 0.30 0.000 CONT 1
1 -6.73 0.001 STOP 1
0 0.30 0.000 CONT 1
0.30 3 0 (GLOBAL E-PARAMETER WITH n OTHER CHOICES, global APW/LAPW)
0 -1.44 0.002 CONT 1
0 0.30 0.000 CONT 1
1 0.30 0.000 CONT 1
K-VECTORS FROM UNIT:4 -12.9 2.5 24 emin/emax/nband #red
SiO2.in2
TOT (TOT,FOR,QTL,EFG,FERMI)
-15.9 22.0 0.50 0.05 EMIN, NE, ESEPERMIN, ESEPER0
GAUSS 0.001 (GAUSS,ROOT,TEMP,TETRA,ALL eval)
0 0 4 0 4 4 6 0 6 4
0 0 4 0 4 4 6 0 6 4 -3 2
14.00 GMAX
NOFILE FILE/NOFILE write recprlist
SiO2_8.0.struct
SiO2-fake fluorite
F LATTICE,NONEQUIV.ATOMS: 2225_Fm-3m
MODE OF CALC=NREL unit=bohr
8.000000 8.000000 8.000000 90.000000 90.000000 90.000000
ATOM 1: X=0.00000000 Y=0.00000000 Z=0.00000000
MULT= 1 ISPLIT= 2
Si NPT= 781 R0=0.00001000 RMT= 1.5000 Z: 14.0
LOCAL ROT MATRIX: 1.0000000 0.0000000 0.0000000
0.0000000 1.0000000 0.0000000
0.0000000 0.0000000 1.0000000
ATOM 2: X=0.25000000 Y=0.25000000 Z=0.25000000
MULT= 2 ISPLIT= 2
2: X=0.75000000 Y=0.75000000 Z=0.75000000
O NPT= 781 R0=0.00001000 RMT= 1.5000 Z: 8.0
LOCAL ROT MATRIX: 1.0000000 0.0000000 0.0000000
0.0000000 1.0000000 0.0000000
0.0000000 0.0000000 1.0000000
48 NUMBER OF SYMMETRY OPERATIONS
1 0 0 0.00000000
0-1 0 0.00000000
0 0-1 0.00000000
1
1 0 0 0.00000000
0 0-1 0.00000000
0-1 0 0.00000000
2
-1 0 0 0.00000000
0-1 0 0.00000000
0 0-1 0.00000000
3
-1 0 0 0.00000000
0 0-1 0.00000000
0-1 0 0.00000000
4
0 1 0 0.00000000
-1 0 0 0.00000000
0 0-1 0.00000000
5
0 0 1 0.00000000
-1 0 0 0.00000000
0-1 0 0.00000000
6
0 1 0 0.00000000
1 0 0 0.00000000
0 0-1 0.00000000
7
0 0 1 0.00000000
1 0 0 0.00000000
0-1 0 0.00000000
8
0 1 0 0.00000000
0 0-1 0.00000000
-1 0 0 0.00000000
9
0 0 1 0.00000000
0-1 0 0.00000000
-1 0 0 0.00000000
10
0 1 0 0.00000000
0 0-1 0.00000000
1 0 0 0.00000000
11
0 0 1 0.00000000
0-1 0 0.00000000
1 0 0 0.00000000
12
0-1 0 0.00000000
-1 0 0 0.00000000
0 0-1 0.00000000
13
0-1 0 0.00000000
1 0 0 0.00000000
0 0-1 0.00000000
14
0 0-1 0.00000000
-1 0 0 0.00000000
0-1 0 0.00000000
15
0 0-1 0.00000000
1 0 0 0.00000000
0-1 0 0.00000000
16
1 0 0 0.00000000
0 1 0 0.00000000
0 0-1 0.00000000
17
-1 0 0 0.00000000
0 1 0 0.00000000
0 0-1 0.00000000
18
1 0 0 0.00000000
0 0 1 0.00000000
0-1 0 0.00000000
19
-1 0 0 0.00000000
0 0 1 0.00000000
0-1 0 0.00000000
20
0-1 0 0.00000000
0 0-1 0.00000000
-1 0 0 0.00000000
21
0 0-1 0.00000000
0-1 0 0.00000000
-1 0 0 0.00000000
22
0-1 0 0.00000000
0 0-1 0.00000000
1 0 0 0.00000000
23
0 0-1 0.00000000
0-1 0 0.00000000
1 0 0 0.00000000
24
0 0 1 0.00000000
0 1 0 0.00000000
-1 0 0 0.00000000
25
0 1 0 0.00000000
0 0 1 0.00000000
-1 0 0 0.00000000
26
0 0 1 0.00000000
0 1 0 0.00000000
1 0 0 0.00000000
27
0 1 0 0.00000000
0 0 1 0.00000000
1 0 0 0.00000000
28
1 0 0 0.00000000
0 0-1 0.00000000
0 1 0 0.00000000
29
-1 0 0 0.00000000
0 0-1 0.00000000
0 1 0 0.00000000
30
1 0 0 0.00000000
0-1 0 0.00000000
0 0 1 0.00000000
31
-1 0 0 0.00000000
0-1 0 0.00000000
0 0 1 0.00000000
32
0 0 1 0.00000000
-1 0 0 0.00000000
0 1 0 0.00000000
33
0 0 1 0.00000000
1 0 0 0.00000000
0 1 0 0.00000000
34
0 1 0 0.00000000
-1 0 0 0.00000000
0 0 1 0.00000000
35
0 1 0 0.00000000
1 0 0 0.00000000
0 0 1 0.00000000
36
0 0-1 0.00000000
0 1 0 0.00000000
-1 0 0 0.00000000
37
0-1 0 0.00000000
0 0 1 0.00000000
-1 0 0 0.00000000
38
0 0-1 0.00000000
0 1 0 0.00000000
1 0 0 0.00000000
39
0-1 0 0.00000000
0 0 1 0.00000000
1 0 0 0.00000000
40
0 0-1 0.00000000
-1 0 0 0.00000000
0 1 0 0.00000000
41
0-1 0 0.00000000
-1 0 0 0.00000000
0 0 1 0.00000000
42
0 0-1 0.00000000
1 0 0 0.00000000
0 1 0 0.00000000
43
0-1 0 0.00000000
1 0 0 0.00000000
0 0 1 0.00000000
44
1 0 0 0.00000000
0 0 1 0.00000000
0 1 0 0.00000000
45
1 0 0 0.00000000
0 1 0 0.00000000
0 0 1 0.00000000
46
-1 0 0 0.00000000
0 0 1 0.00000000
0 1 0 0.00000000
47
-1 0 0 0.00000000
0 1 0 0.00000000
0 0 1 0.00000000
48
optimize.job
#!/bin/csh -f
# Modify this script according to your needs:
# Uncomment one of the lines ...
# Change run_lapw to runsp_lapw or use different convergence criterium
# Change save_lapw -d XXX
foreach i ( \
SiO2_08.0\
SiO2_08.1\
SiO2_08.2\
SiO2_08.3\
SiO2_08.4\
SiO2_08.5\
SiO2_08.6\
SiO2_08.7\
SiO2_08.8\
SiO2_08.9\
SiO2_09.0\
)
rm SiO2.struct # NFS-bug
cp $i.struct SiO2.struct
# Please uncomment and adapt any of the lines below according to your needs
# cp $i.clmsum C.clmsum
x dstart
# x dstart -c
# run_lapw -ec 0.0001 -in1new 3 -in1orig -renorm
# runsp_lapw -ec 0.0001
# min -I -j "run_lapw -I -fc 1.0 -i 40 "
run_lapw -ec 0.000001
set stat = $status
if ($stat) then
echo "ERROR status in" $i
exit 1
endif
save_lapw $i
# save_lapw -f -d XXX $i
end
Thanks for your suggestion. For exciting, I used the species file from the download tar file which I believe is the same as the one on line. For wien2k, I have attached some of the input files. A modified optimize script was used to run with the same lattice constants as for exciting. thanks again for your feedback. Natalie Holzwarth Actually -- google would not let me attach the files so I list them below
SiO2.in0
TOT 5 (5...CA-LDA, 13...PBE-GGA, 11...WC-GGA)
NR2V IFFT (R2V)
36 36 36 2.00 1 min IFFT-parameters, enhancement factor, iprint
SiO2.in1
WFFIL EF=.56485 (WFFIL, WFPRI, ENFIL, SUPWF)
9.00 12 6 (R-MT*K-MAX; MAX L IN WF, V-NMT
0.30 3 0 (GLOBAL E-PARAMETER WITH n OTHER CHOICES, global APW/LAPW)
1 0.30 0.000 CONT 1
1 -6.73 0.001 STOP 1
0 0.30 0.000 CONT 1
0.30 3 0 (GLOBAL E-PARAMETER WITH n OTHER CHOICES, global APW/LAPW)
0 -1.44 0.002 CONT 1
0 0.30 0.000 CONT 1
1 0.30 0.000 CONT 1
K-VECTORS FROM UNIT:4 -12.9 2.5 24 emin/emax/nband #red
SiO2.in2
TOT (TOT,FOR,QTL,EFG,FERMI)
-15.9 22.0 0.50 0.05 EMIN, NE, ESEPERMIN, ESEPER0
GAUSS 0.001 (GAUSS,ROOT,TEMP,TETRA,ALL eval)
0 0 4 0 4 4 6 0 6 4
0 0 4 0 4 4 6 0 6 4 -3 2
14.00 GMAX
NOFILE FILE/NOFILE write recprlist
SiO2_8.0.struct
SiO2-fake fluorite
F LATTICE,NONEQUIV.ATOMS: 2225_Fm-3m
MODE OF CALC=NREL unit=bohr
8.000000 8.000000 8.000000 90.000000 90.000000 90.000000
ATOM 1: X=0.00000000 Y=0.00000000 Z=0.00000000
MULT= 1 ISPLIT= 2
Si NPT= 781 R0=0.00001000 RMT= 1.5000 Z: 14.0
LOCAL ROT MATRIX: 1.0000000 0.0000000 0.0000000
0.0000000 1.0000000 0.0000000
0.0000000 0.0000000 1.0000000
ATOM 2: X=0.25000000 Y=0.25000000 Z=0.25000000
MULT= 2 ISPLIT= 2
2: X=0.75000000 Y=0.75000000 Z=0.75000000
O NPT= 781 R0=0.00001000 RMT= 1.5000 Z: 8.0
LOCAL ROT MATRIX: 1.0000000 0.0000000 0.0000000
0.0000000 1.0000000 0.0000000
0.0000000 0.0000000 1.0000000
48 NUMBER OF SYMMETRY OPERATIONS
1 0 0 0.00000000
0-1 0 0.00000000
0 0-1 0.00000000
1
1 0 0 0.00000000
0 0-1 0.00000000
0-1 0 0.00000000
2
-1 0 0 0.00000000
0-1 0 0.00000000
0 0-1 0.00000000
3
-1 0 0 0.00000000
0 0-1 0.00000000
0-1 0 0.00000000
4
0 1 0 0.00000000
-1 0 0 0.00000000
0 0-1 0.00000000
5
0 0 1 0.00000000
-1 0 0 0.00000000
0-1 0 0.00000000
6
0 1 0 0.00000000
1 0 0 0.00000000
0 0-1 0.00000000
7
0 0 1 0.00000000
1 0 0 0.00000000
0-1 0 0.00000000
8
0 1 0 0.00000000
0 0-1 0.00000000
-1 0 0 0.00000000
9
0 0 1 0.00000000
0-1 0 0.00000000
-1 0 0 0.00000000
10
0 1 0 0.00000000
0 0-1 0.00000000
1 0 0 0.00000000
11
0 0 1 0.00000000
0-1 0 0.00000000
1 0 0 0.00000000
12
0-1 0 0.00000000
-1 0 0 0.00000000
0 0-1 0.00000000
13
0-1 0 0.00000000
1 0 0 0.00000000
0 0-1 0.00000000
14
0 0-1 0.00000000
-1 0 0 0.00000000
0-1 0 0.00000000
15
0 0-1 0.00000000
1 0 0 0.00000000
0-1 0 0.00000000
16
1 0 0 0.00000000
0 1 0 0.00000000
0 0-1 0.00000000
17
-1 0 0 0.00000000
0 1 0 0.00000000
0 0-1 0.00000000
18
1 0 0 0.00000000
0 0 1 0.00000000
0-1 0 0.00000000
19
-1 0 0 0.00000000
0 0 1 0.00000000
0-1 0 0.00000000
20
0-1 0 0.00000000
0 0-1 0.00000000
-1 0 0 0.00000000
21
0 0-1 0.00000000
0-1 0 0.00000000
-1 0 0 0.00000000
22
0-1 0 0.00000000
0 0-1 0.00000000
1 0 0 0.00000000
23
0 0-1 0.00000000
0-1 0 0.00000000
1 0 0 0.00000000
24
0 0 1 0.00000000
0 1 0 0.00000000
-1 0 0 0.00000000
25
0 1 0 0.00000000
0 0 1 0.00000000
-1 0 0 0.00000000
26
0 0 1 0.00000000
0 1 0 0.00000000
1 0 0 0.00000000
27
0 1 0 0.00000000
0 0 1 0.00000000
1 0 0 0.00000000
28
1 0 0 0.00000000
0 0-1 0.00000000
0 1 0 0.00000000
29
-1 0 0 0.00000000
0 0-1 0.00000000
0 1 0 0.00000000
30
1 0 0 0.00000000
0-1 0 0.00000000
0 0 1 0.00000000
31
-1 0 0 0.00000000
0-1 0 0.00000000
0 0 1 0.00000000
32
0 0 1 0.00000000
-1 0 0 0.00000000
0 1 0 0.00000000
33
0 0 1 0.00000000
1 0 0 0.00000000
0 1 0 0.00000000
34
0 1 0 0.00000000
-1 0 0 0.00000000
0 0 1 0.00000000
35
0 1 0 0.00000000
1 0 0 0.00000000
0 0 1 0.00000000
36
0 0-1 0.00000000
0 1 0 0.00000000
-1 0 0 0.00000000
37
0-1 0 0.00000000
0 0 1 0.00000000
-1 0 0 0.00000000
38
0 0-1 0.00000000
0 1 0 0.00000000
1 0 0 0.00000000
39
0-1 0 0.00000000
0 0 1 0.00000000
1 0 0 0.00000000
40
0 0-1 0.00000000
-1 0 0 0.00000000
0 1 0 0.00000000
41
0-1 0 0.00000000
-1 0 0 0.00000000
0 0 1 0.00000000
42
0 0-1 0.00000000
1 0 0 0.00000000
0 1 0 0.00000000
43
0-1 0 0.00000000
1 0 0 0.00000000
0 0 1 0.00000000
44
1 0 0 0.00000000
0 0 1 0.00000000
0 1 0 0.00000000
45
1 0 0 0.00000000
0 1 0 0.00000000
0 0 1 0.00000000
46
-1 0 0 0.00000000
0 0 1 0.00000000
0 1 0 0.00000000
47
-1 0 0 0.00000000
0 1 0 0.00000000
0 0 1 0.00000000
48
optimize.job
#!/bin/csh -f
# Modify this script according to your needs:
# Uncomment one of the lines ...
# Change run_lapw to runsp_lapw or use different convergence criterium
# Change save_lapw -d XXX
foreach i ( \
SiO2_08.0\
SiO2_08.1\
SiO2_08.2\
SiO2_08.3\
SiO2_08.4\
SiO2_08.5\
SiO2_08.6\
SiO2_08.7\
SiO2_08.8\
SiO2_08.9\
SiO2_09.0\
)
rm SiO2.struct # NFS-bug
cp $i.struct SiO2.struct
# Please uncomment and adapt any of the lines below according to your needs
# cp $i.clmsum C.clmsum
x dstart
# x dstart -c
# run_lapw -ec 0.0001 -in1new 3 -in1orig -renorm
# runsp_lapw -ec 0.0001
# min -I -j "run_lapw -I -fc 1.0 -i 40 "
run_lapw -ec 0.000001
set stat = $status
if ($stat) then
echo "ERROR status in" $i
exit 1
endif
save_lapw $i
# save_lapw -f -d XXX $i
end
Reply all
Reply to author
Forward
0 new messages