SGCPMD: problem with equilibration?
291 views
Skip to first unread message

Alexandra Dávila
Sep 15, 2022, 6:04:35 AM9/15/22
to cp2k
Dear Cp2k users,
I have carried on a
second-generation Car-Parrinello (SGCP) MD simulation for Pt(111)/water
interface. Unit cell contains Pt: 6x6x4 (2 frozen layers), water: 134
molecules.
I have performed a similar workflow as Onofrio ("SGCPMD: number of steps per SCF run increase suddenly after initial 400 MD steps"). But I have used the NOISY_GAMMA as in the publication "First-Principles Simulations of an Aqueous CO/Pt(111) Interface" doi:10.1021/acs.jpcc.8b05933, Pt: 5E-05 1/fs and for water 2.2E-04 1/fs. Temperature is set to 330K.
Unfortunately,
in my case along the SGMD the temperature of Pt region drops to roughly
180 K, the temperature of the hole system is about 328K. I expected
that the temperature of Pt region would be closer to the water after a
equilibration time of 4ps. Does this mean that probably my system is not
well-equilibrated? Is this going to affect the dynamics and energy of the system?
(PS: the density profile seems ok, double peak on oxygen, well-known for
Pt(111))
Many thanks in advance!
Alexandra
PS: I also observed the increase of the SCF steps after a restart!
TOP: red dashed line: average temperature of system
BOTTOM: temperature for each region
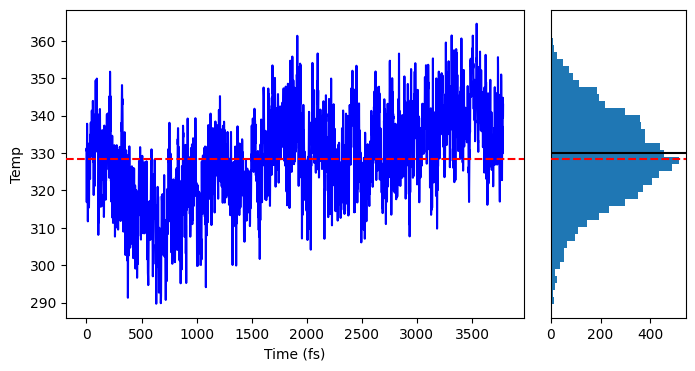
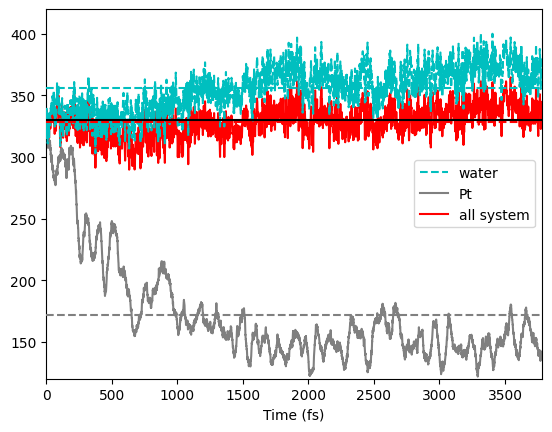

Onofrio Tau
Sep 15, 2022, 6:53:45 AM9/15/22
to cp2k
Dear Alexandra,
have you done an equilibration step before starting the SGCPMD run?
As I can see from your plots it seems that it is not equilibrated yet. I suggest you to make a BOMD run first for better equilibration, then switching to sgcpmd, as follows:
1. BOMD as NVE ensemble + TEMP_TOL 100
2. switch to SGCPMD using RESTART.wfn of the previous equilibration step as a wfn guess
It works for me.
Hope it helps
Regards
Onofrio
Alexandra Dávila
Sep 15, 2022, 7:34:39 AM9/15/22
to cp2k
Dear Onofrio,
thanks a lot for your fast response! Indeed, I did a NVE simulation with Langevin (as suggested here:https://www.cp2k.org/howto:md). Then I used the geometry and wfn for the SGCPMD.
As you, I use TEMP_TOL, but I set it to the value of 50 for each region. Do you think that it makes the difference?
This are the parameters that I used for the NVE:
&MD
ENSEMBLE LANGEVIN
STEPS 6000
TIMESTEP 5.0E-01
TEMPERATURE 3.3000000000000006E+02
TEMP_KIND T
SCALE_TEMP_KIND T
&LANGEVIN
GAMMA 1.0000000000000000E-03
NOISY_GAMMA 0
&END LANGEVIN
&THERMAL_REGION
DO_LANGEVIN_DEFAULT F
&DEFINE_REGION
DO_LANGEVIN F
LIST 145..546
TEMPERATURE 3.3000000000000006E+02
NOISY_GAMMA_REGION 2.1999999999999998E-04
TEMP_TOL 50
&END DEFINE_REGION
&DEFINE_REGION
DO_LANGEVIN F
LIST 73..144
TEMPERATURE 3.3000000000000006E+02
NOISY_GAMMA_REGION 4.9999999999999996E-05
TEMP_TOL 50
&END DEFINE_REGION
&END THERMAL_REGION
&END MD
ENSEMBLE LANGEVIN
STEPS 6000
TIMESTEP 5.0E-01
TEMPERATURE 3.3000000000000006E+02
TEMP_KIND T
SCALE_TEMP_KIND T
&LANGEVIN
GAMMA 1.0000000000000000E-03
NOISY_GAMMA 0
&END LANGEVIN
&THERMAL_REGION
DO_LANGEVIN_DEFAULT F
&DEFINE_REGION
DO_LANGEVIN F
LIST 145..546
TEMPERATURE 3.3000000000000006E+02
NOISY_GAMMA_REGION 2.1999999999999998E-04
TEMP_TOL 50
&END DEFINE_REGION
&DEFINE_REGION
DO_LANGEVIN F
LIST 73..144
TEMPERATURE 3.3000000000000006E+02
NOISY_GAMMA_REGION 4.9999999999999996E-05
TEMP_TOL 50
&END DEFINE_REGION
&END THERMAL_REGION
&END MD
The NOISY_GAMMA is not considered in the simulation. Is there something not so good with the set of parameters I used?
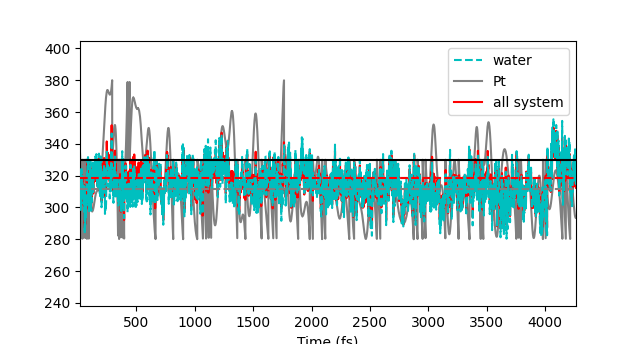
I appreciate a lot your feedback!
Best regards,
Aexandra
Onofrio Tau
Sep 15, 2022, 8:04:00 AM9/15/22
to cp2k
Dear Alexandra,
I used to do ENSEMBLE NVE in &MD for the BOMD run. Probably 50 K of the TEMP_TOL is too strict, at least for Pt. As you can see from your last plot, the Temperature of the Pt region frequently drops to your lower limit, 280 K, even after 4000 fs (not good). This is why you get a Pt temperature drop in the SGCPMD run, because of a not good previous equilibration.Regards.
Onofrio
Alexandra Dávila
Sep 15, 2022, 2:11:01 PM9/15/22
to cp2k
Dear Onofrio!
you are right! It is too tight. I will try out what you recommende me!
Thanks a lot,
Alexandra
El jueves, 15 de septiembre de 2022 a las 12:53:45 UTC+2, onofr...@gmail.com escribió:
Onofrio Tau
Sep 16, 2022, 4:03:11 AM9/16/22
to cp2k
Let me know if it works for your system. Can I ask you how many steps per SCF run are tipically required for convergence in your SGCPMD run?
Thanks.
Onofrio
Alexandra Dávila
Sep 16, 2022, 6:42:10 AM9/16/22
to cp2k
I will!
After a restart, I get 3-6 steps, but then it goes high to ~15. Sometimes it could be higher, but then goes again down.
Resgards,
Alexandra
Reply all
Reply to author
Forward
0 new messages