WARNING in particle_methods.F:684 :: The distance between the atoms *** *** 10 and 405 is only 0.496 angstrom and thus smaller than the threshold *** *** of 0.500 angstrom
254 views
Skip to first unread message

Jessie wang
Jul 7, 2022, 5:22:07 AM7/7/22
to cp2k
Dear all,
The initial geometry is actually the result of Gaussian's geometry optimization. So the initial input of the structure should be reasonable enough for cp2k to run and converge in a short time.
I am new to cp2k and am performing a test geometry optimization.
The initial geometry is actually the result of Gaussian's geometry optimization. So the initial input of the structure should be reasonable enough for cp2k to run and converge in a short time.
But it displays "WARNING in particle_methods.F:684 :: The distance between the atoms *** *** 10 and 405 is only 0.496 angstrom smaller than the threshold *** *** of 0.500 angstrom " a lot. And the program seems to be stuck in a weird corner and not updating anything in the output, but it keeps running on the server. I here attached the inp and out file here.
I'm appreciate it if you can help me with this or provide any information refers to the problem.
Thank you !!

Marcella Iannuzzi
Jul 7, 2022, 5:55:36 AM7/7/22
to cp2k
Hi
The simulation CELL is most probably far too small for the system.
Using
&POISSON
PERIODIC NONE
POISSON_SOLVER MT
&MT
ALPHA 7.0
REL_CUTOFF 1.2
&END MT
&END POISSON
PERIODIC NONE
POISSON_SOLVER MT
&MT
ALPHA 7.0
REL_CUTOFF 1.2
&END MT
&END POISSON
a CELL that is approximately twice the size of the system in all direction is recommended.
Regards
Marcella

Anna Hehn
Jul 7, 2022, 6:00:45 AM7/7/22
to cp...@googlegroups.com
Dear Jessie,
maybe the coordinates that you give as input are not in the correct units (angstrom / bohr). You also don't specify the units in the cell section.
I would check this first to make sure that the cell vectors and the coordinates have the correct units.
Best regards
Anna
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/955838ca-3cee-489d-9ca8-90f0bae8ad9an%40googlegroups.com.
Anna Hehn
Jul 7, 2022, 6:02:41 AM7/7/22
to cp...@googlegroups.com
Dear Jessie,
I saw Marcella's response only after typing, so stick with hers.
Best regards,
Anna
Jessie wang
Jul 7, 2022, 6:06:40 AM7/7/22
to cp2k
Thank you very much for the advice.
I am using POISSON since it is an isolated cluster and a non-periodic system.
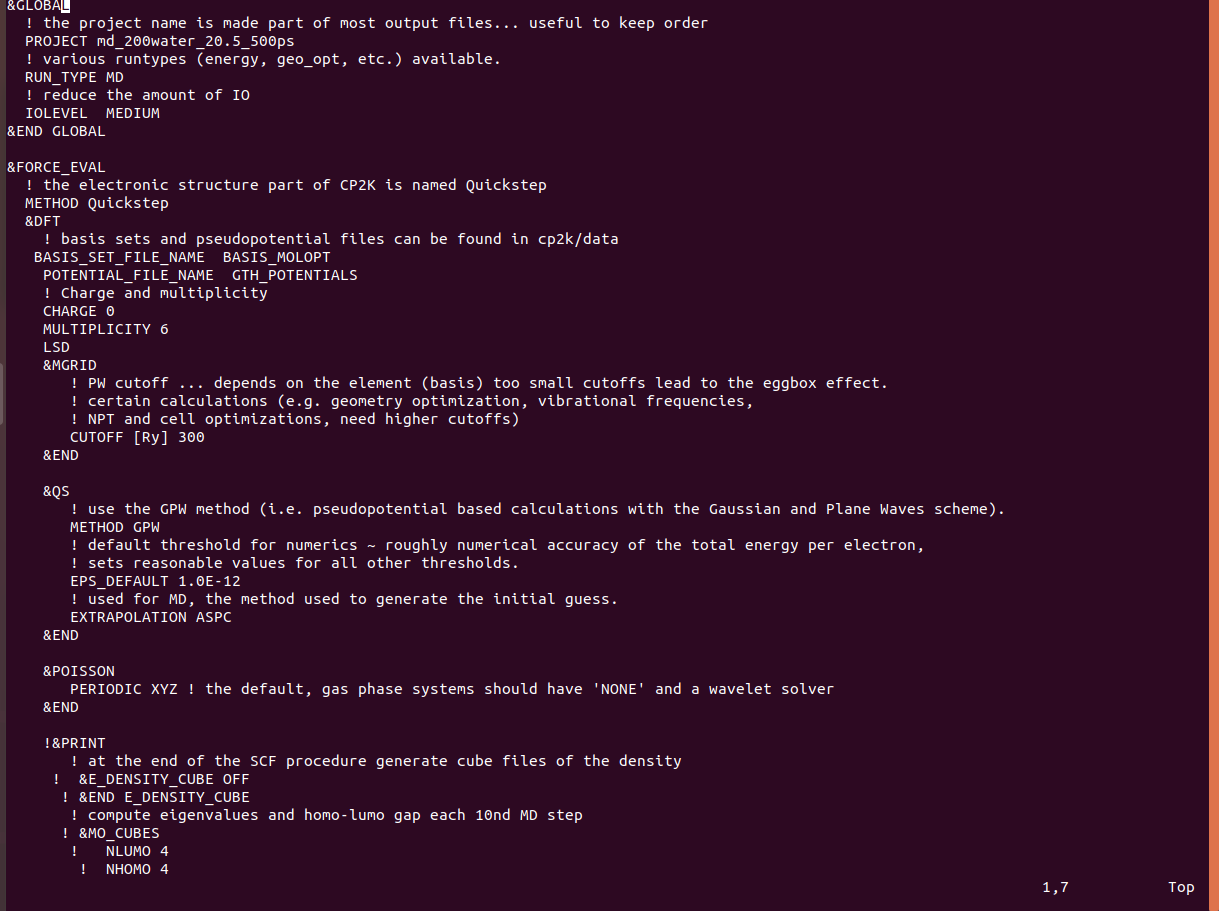
I did not specify any cell parameters in the input file since it is a cluster. I don't think there is any reason to specify a cell, and I believe the cp2k manual indicates that the program defaults to considering the project an isolated system if there are no cell parameters. Does this make sense to you?
It is true that cp2k terminates the program by itself if there are no cell parameters for MD, but for Geo, cp2k just keeps running without any error message.
I am using POISSON since it is an isolated cluster and a non-periodic system.
I did not specify any cell parameters in the input file since it is a cluster. I don't think there is any reason to specify a cell, and I believe the cp2k manual indicates that the program defaults to considering the project an isolated system if there are no cell parameters. Does this make sense to you?
It is true that cp2k terminates the program by itself if there are no cell parameters for MD, but for Geo, cp2k just keeps running without any error message.
Jessie wang
Jul 7, 2022, 6:08:25 AM7/7/22
to cp2k
Thank you Anna, your advice is very valuable. I'm going to check the units right now !
Jessie wang
Jul 7, 2022, 6:19:41 AM7/7/22
to cp2k
Checked the unit, not the problem.
T^T
Marcella Iannuzzi
Jul 7, 2022, 6:21:23 AM7/7/22
to cp2k
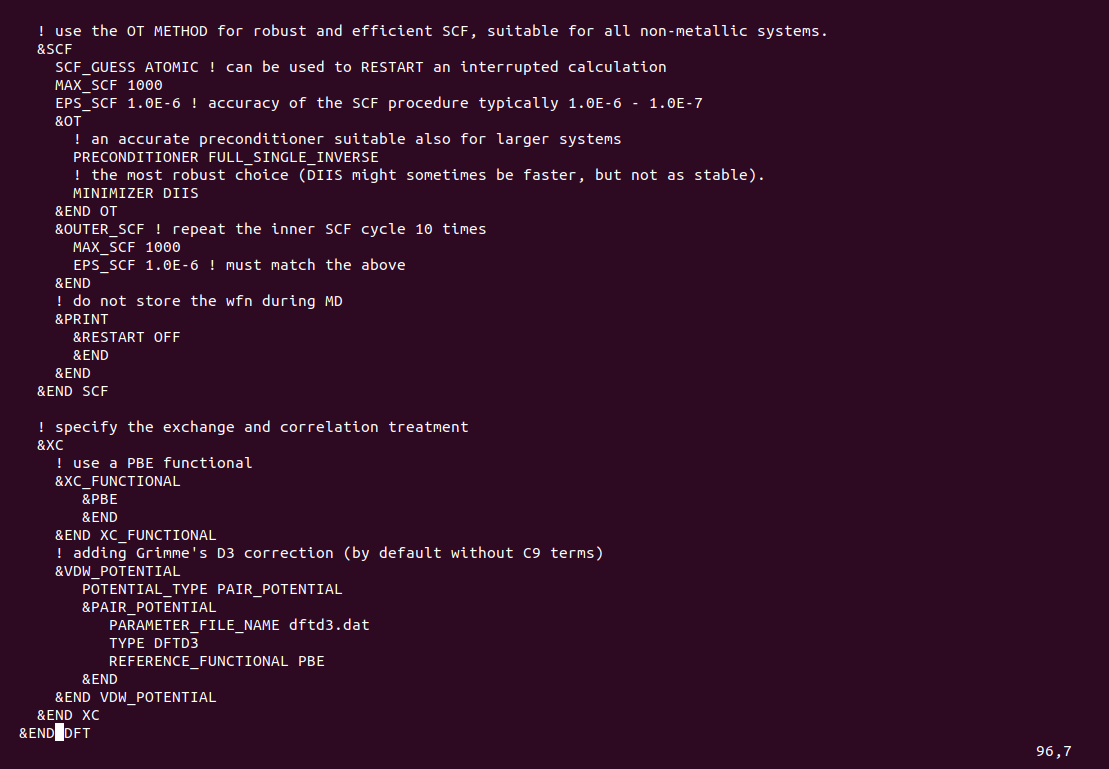
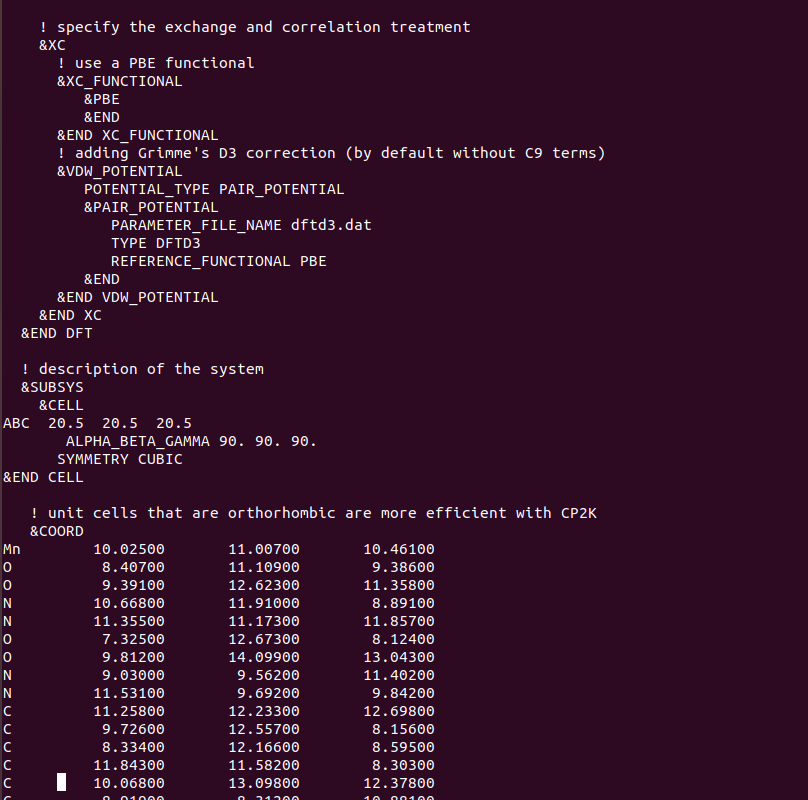
You have specified the CELL parameters
&CELL
ABC 8.1300 5.1320 3.1302
ALPHA_BETA_GAMMA 90 90 90
&END CELL
What you have not done is to set also for the CELL
PERIODIC NONE, since the default is XYZ
This is a problem
The CELL parameters are always needed also in the case of PERIODIC NONE
in particular, enough vacuum space around the molecular system is required such that the selected Poisson solver for non periodic conditions works correctly
Regards
Marcella

{kind=link}
{kind=link}
{kind=link}
0 new messages