Not getting correct optimised geometry for Au(111) surface with ligands (BDT or DMBT)
162 views
Skip to first unread message

Vikash Khokhar
Nov 28, 2022, 7:58:13 AM11/28/22
to cp2k
I'm trying to optimize a small unit cell of Au(111) surface with ligands (BDT or DMBT) but I'm not getting the correct optimized geometry.
I'm using Cp2k 8.1 version.
I'm attaching the Input file here, please have a look and suggest to me what's wrong with my calculation.
&GLOBAL
PROJECT Au_64_2BDT
RUN_TYPE GEO_OPT
PRINT_LEVEL MEDIUM
&END GLOBAL
&MOTION
&GEO_OPT
TYPE MINIMIZATION
OPTIMIZER BFGS
MAX_DR 3.00E-03
MAX_FORCE 4.50E-04
RMS_DR 1.50E-03
RMS_FORCE 3.00E-04
MAX_ITER 3000
&END GEO_OPT
&CONSTRAINT
&FIXED_ATOMS
COMPONENTS_TO_FIX XYZ
LIST 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
&END FIXED_ATOMS
&END CONSTRAINT
&END MOTION
&FORCE_EVAL
METHOD QS
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
&QS
METHOD GPW
EXTRAPOLATION USE_GUESS
EPS_DEFAULT 1.0E-10
&END QS
&MGRID
CUTOFF 350
NGRIDS 4
REL_CUTOFF 60
&END MGRID
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-05
MAX_SCF 200
ADDED_MOS 10
&OUTER_SCF
EPS_SCF 1.0E-04
MAX_SCF 200
&END OUTER_SCF
&DIAGONALIZATION T
ALGORITHM STANDARD
&END DIAGONALIZATION
&SMEAR ON
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE [K] 300
&END SMEAR
&MIXING T
METHOD BROYDEN_MIXING
ALPHA 0.4
NBROYDEN 8
&END MIXING
&END SCF
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&vdW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
PARAMETER_FILE_NAME dftd3.dat
TYPE DFTD3
REFERENCE_FUNCTIONAL PBE
R_CUTOFF 25.0
&END PAIR_POTENTIAL
&END vdW_POTENTIAL
&END XC
&KPOINTS
SCHEME GAMMA 1 1 1
FULL_GRID .TRUE.
PARALLEL_GROUP_SIZE 0
&END KPOINTS
&END DFT
&SUBSYS
&CELL
PERIODIC XYZ
A 17.3099747000000015 0.0000000000000000 0.0000000000000000
B 0.0000000000000000 9.9939184000000001 0.0000000000000000
C 0.0000000000000000 0.0000000000000000 30.0000000000000000
&END CELL
&COORD
@INCLUDE Au_64_2BDT.xyz
&END COORD
&KIND Au
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE
&END KIND
&KIND H
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-PBE
&END KIND
&KIND C
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-PBE
&END KIND
&KIND S
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-PBE
&END KIND
&END SUBSYS
&END FORCE_EVAL
PROJECT Au_64_2BDT
RUN_TYPE GEO_OPT
PRINT_LEVEL MEDIUM
&END GLOBAL
&MOTION
&GEO_OPT
TYPE MINIMIZATION
OPTIMIZER BFGS
MAX_DR 3.00E-03
MAX_FORCE 4.50E-04
RMS_DR 1.50E-03
RMS_FORCE 3.00E-04
MAX_ITER 3000
&END GEO_OPT
&CONSTRAINT
&FIXED_ATOMS
COMPONENTS_TO_FIX XYZ
LIST 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
&END FIXED_ATOMS
&END CONSTRAINT
&END MOTION
&FORCE_EVAL
METHOD QS
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
&QS
METHOD GPW
EXTRAPOLATION USE_GUESS
EPS_DEFAULT 1.0E-10
&END QS
&MGRID
CUTOFF 350
NGRIDS 4
REL_CUTOFF 60
&END MGRID
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-05
MAX_SCF 200
ADDED_MOS 10
&OUTER_SCF
EPS_SCF 1.0E-04
MAX_SCF 200
&END OUTER_SCF
&DIAGONALIZATION T
ALGORITHM STANDARD
&END DIAGONALIZATION
&SMEAR ON
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE [K] 300
&END SMEAR
&MIXING T
METHOD BROYDEN_MIXING
ALPHA 0.4
NBROYDEN 8
&END MIXING
&END SCF
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&vdW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
PARAMETER_FILE_NAME dftd3.dat
TYPE DFTD3
REFERENCE_FUNCTIONAL PBE
R_CUTOFF 25.0
&END PAIR_POTENTIAL
&END vdW_POTENTIAL
&END XC
&KPOINTS
SCHEME GAMMA 1 1 1
FULL_GRID .TRUE.
PARALLEL_GROUP_SIZE 0
&END KPOINTS
&END DFT
&SUBSYS
&CELL
PERIODIC XYZ
A 17.3099747000000015 0.0000000000000000 0.0000000000000000
B 0.0000000000000000 9.9939184000000001 0.0000000000000000
C 0.0000000000000000 0.0000000000000000 30.0000000000000000
&END CELL
&COORD
@INCLUDE Au_64_2BDT.xyz
&END COORD
&KIND Au
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE
&END KIND
&KIND H
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-PBE
&END KIND
&KIND C
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-PBE
&END KIND
&KIND S
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-PBE
&END KIND
&END SUBSYS
&END FORCE_EVAL
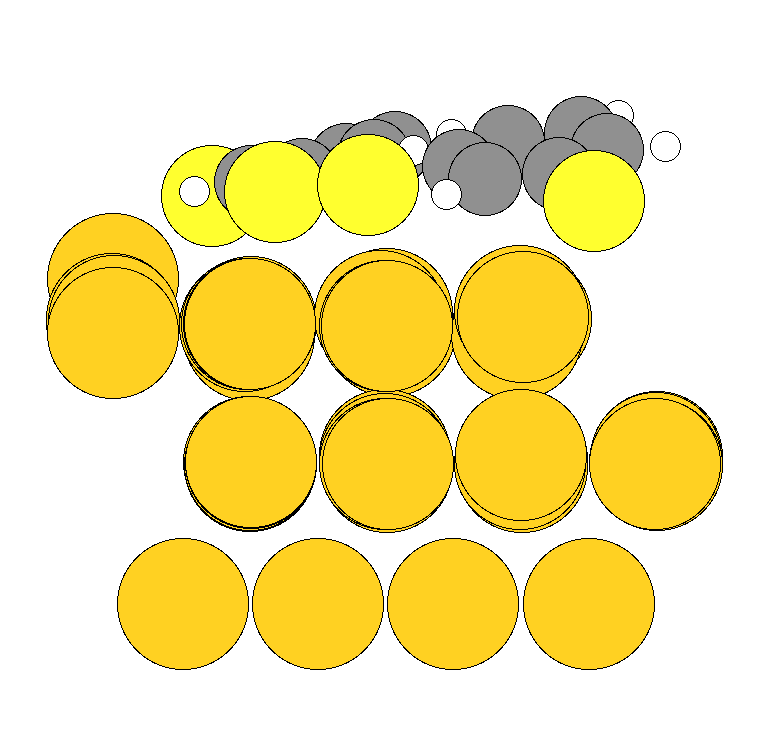
I'm also attaching the input and output geometry pics. In the input 3rd layer of the gold surface is fixed and the above 2 layers optimize above each other(which breaks 111 symmetry) which is not expected.
I hope you understand my problem and I expect a solution, earliest possible.
Regards
Vikash Khokhar
Indian Institute of Technology, Madras 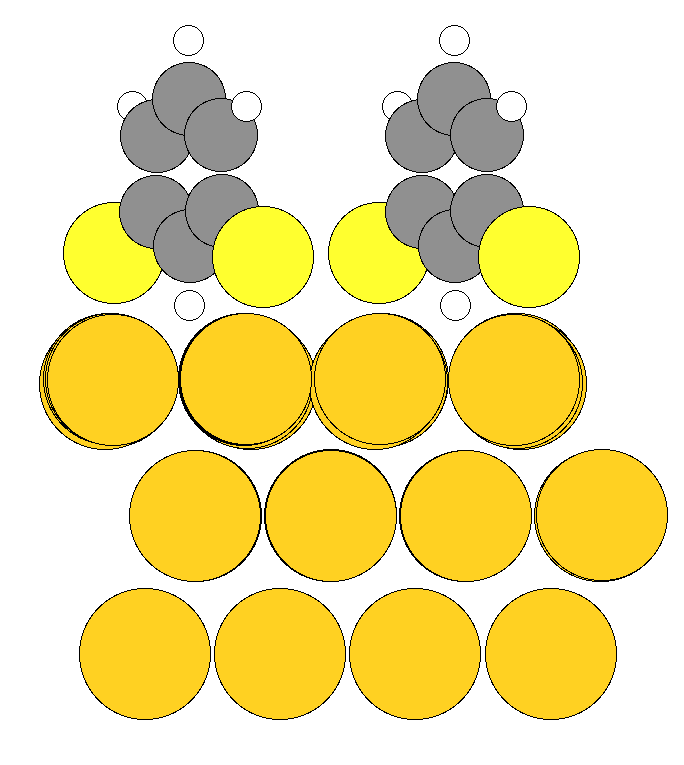

Marcella Iannuzzi
Nov 28, 2022, 9:47:19 AM11/28/22
to cp2k
dear Vikash
The system is probably too small.
To stabilise the Au slab one should use more layers, we use 6.
With such small lateral dimension you should use a mesh of k-points.
Moreover, the tolerance for the convergence of the wavefunction is not very tight and the PW cutoff seems quite low.
Regards
Marcella
Vikash Khokhar
Nov 28, 2022, 11:58:54 PM11/28/22
to cp2k
Thank you, Marcella,
I would like to know the exact PW cutoff I can use.
And yes, I have used mesh k-points 2 3 1, are these correct, or do I need to take a bigger grid? If yes, what should it be?
I thought the default tolerance would be okay. If not, then how much tolerance would be enough?
I would like to know the exact PW cutoff I can use.
And yes, I have used mesh k-points 2 3 1, are these correct, or do I need to take a bigger grid? If yes, what should it be?
I thought the default tolerance would be okay. If not, then how much tolerance would be enough?
Regards
Vikash
Vikash Khokhar
Nov 29, 2022, 12:14:59 AM11/29/22
to cp2k
And one more thing. I only need the correct geometry. Energetics or forces are not required at the moment.
Marcella Iannuzzi
Nov 29, 2022, 7:15:58 AM11/29/22
to cp2k
Dear Vikash
In the posted input, the k-points are not used.
If you already used them, try with a denser mesh and with the MONKHORST-PACK scheme.
I suggest that you test on one configuration (possibly already a bit distorted) the effects on energies and forces by changing k-point mesh and cutoff.
If energies and forces are not accurate, the geometry will be wrong. EPS_SCF 1.0E-07 should be good enough.
OUTER_SCF is used only to reinitialise the preconditioner for the OT approach, hence it is useless with the standard diagonalization method.
3 layers for a Au slab are insufficient and are keen to lead to weird distortions in the lattice.
Regards
Marcella
Vikash Khokhar
Nov 29, 2022, 7:22:42 AM11/29/22
to cp2k
Thank you, Marcella
I'll look into this.
Regards
I'll look into this.
Regards
Vikash
Reply all
Reply to author
Forward
0 new messages