Second generation Car–Parrinello molecular dynamics
213 views
Skip to first unread message

DMITRII Drugov
Nov 25, 2022, 2:09:07 AM11/25/22
to cp2k
Dear CP2k community,
I am trying to run the 2nd order CPMD. I have ionic liquid system in NVT ensemble.
Could you please advice how to select GAMMA and NOISY_GAMMA?
Also in this section below.
&MOTION
&MD
ENSEMBLE LANGEVIN
...
&LANGEVIN
GAMMA 0.001
&END LANGEVIN
...
&END MD
&MD
ENSEMBLE LANGEVIN
...
&LANGEVIN
GAMMA 0.001
&END LANGEVIN
...
&END MD
I have my BOMD script below.
Many thanks,
Dmitrii
&FORCE_EVAL
METHOD QS
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
CHARGE 0
MULTIPLICITY 1
&MGRID
CUTOFF 400
NGRIDS 4
REL_CUTOFF 40
&END MGRID
&QS
METHOD GAPW
EPS_DEFAULT 1.0E-12
EXTRAPOLATION ASPC
EXTRAPOLATION_ORDER 3
&END
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-6
MAX_SCF 15
&OT
MINIMIZER DIIS
PRECONDITIONER FULL_KINETIC
&END OT
&OUTER_SCF
EPS_SCF 1.0E-6
MAX_SCF 20
&END
&PRINT
&RESTART
&EACH
MD 0
&END EACH
&END
&END
&END SCF
&XC
&XC_FUNCTIONAL BLYP
&END XC_FUNCTIONAL
&XC_GRID
XC_DERIV NN10_SMOOTH
XC_SMOOTH_RHO NN10
&END XC_GRID
&vdW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
PARAMETER_FILE_NAME dftd3.dat
TYPE DFTD3
REFERENCE_FUNCTIONAL BLYP
&END PAIR_POTENTIAL
&END vdW_POTENTIAL
&END XC
&POISSON
PERIODIC xyz
POISSON_SOLVER PERIODIC
&END POISSON
&END DFT
&SUBSYS
&CELL
ABC 19.3457 19.3457 19.3457
PERIODIC xyz
&END CELL
&TOPOLOGY
COORD_FILE_NAME npt.xyz
COORD_FILE_FORMAT XYZ
&END TOPOLOGY
&KIND H
BASIS_SET TZV2P-MOLOPT-GTH
POTENTIAL GTH-BLYP-q1
&END KIND
&KIND Al
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-BLYP-q3
&END KIND
&KIND F
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q7
&END KIND
&KIND O
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q6
&END KIND
&KIND C
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q4
&END KIND
&KIND S
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q6
&END KIND
&KIND N
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q5
&END KIND
&END SUBSYS
&END FORCE_EVAL
&GLOBAL
PROJECT IL_nvt
RUN_TYPE MD
PRINT_LEVEL LOW
FFTW_PLAN_TYPE EXHAUSTIVE
&END GLOBAL
&MOTION
&MD
ENSEMBLE NVT
STEPS 2000
TIMESTEP 0.5
&THERMOSTAT
TYPE NOSE
REGION MASSIVE
&NOSE
TIMECON 10.00
&END NOSE
&END THERMOSTAT
TEMPERATURE 303
&END MD
&PRINT
&TRAJECTORY
&EACH
MD 1
&END EACH
&END TRAJECTORY
&VELOCITIES OFF
&END VELOCITIES
&FORCES OFF
&END FORCES
&RESTART_HISTORY
&EACH
MD 500
&END EACH
&END RESTART_HISTORY
&RESTART
BACKUP_COPIES 3
&EACH
MD 1
&END EACH
&END RESTART
&END PRINT
&END
&EXT_RESTART
RESTART_FILE_NAME nvt-1.restart
&END
METHOD QS
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
CHARGE 0
MULTIPLICITY 1
&MGRID
CUTOFF 400
NGRIDS 4
REL_CUTOFF 40
&END MGRID
&QS
METHOD GAPW
EPS_DEFAULT 1.0E-12
EXTRAPOLATION ASPC
EXTRAPOLATION_ORDER 3
&END
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-6
MAX_SCF 15
&OT
MINIMIZER DIIS
PRECONDITIONER FULL_KINETIC
&END OT
&OUTER_SCF
EPS_SCF 1.0E-6
MAX_SCF 20
&END
&RESTART
&EACH
MD 0
&END EACH
&END
&END
&END SCF
&XC
&XC_FUNCTIONAL BLYP
&END XC_FUNCTIONAL
&XC_GRID
XC_DERIV NN10_SMOOTH
XC_SMOOTH_RHO NN10
&END XC_GRID
&vdW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
PARAMETER_FILE_NAME dftd3.dat
TYPE DFTD3
REFERENCE_FUNCTIONAL BLYP
&END PAIR_POTENTIAL
&END vdW_POTENTIAL
&END XC
&POISSON
PERIODIC xyz
POISSON_SOLVER PERIODIC
&END POISSON
&END DFT
&SUBSYS
&CELL
ABC 19.3457 19.3457 19.3457
PERIODIC xyz
&END CELL
&TOPOLOGY
COORD_FILE_NAME npt.xyz
COORD_FILE_FORMAT XYZ
&END TOPOLOGY
&KIND H
BASIS_SET TZV2P-MOLOPT-GTH
POTENTIAL GTH-BLYP-q1
&END KIND
&KIND Al
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-BLYP-q3
&END KIND
&KIND F
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q7
&END KIND
&KIND O
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q6
&END KIND
&KIND C
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q4
&END KIND
&KIND S
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q6
&END KIND
&KIND N
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q5
&END KIND
&END SUBSYS
&END FORCE_EVAL
&GLOBAL
PROJECT IL_nvt
RUN_TYPE MD
PRINT_LEVEL LOW
FFTW_PLAN_TYPE EXHAUSTIVE
&END GLOBAL
&MOTION
&MD
ENSEMBLE NVT
STEPS 2000
TIMESTEP 0.5
&THERMOSTAT
TYPE NOSE
REGION MASSIVE
&NOSE
TIMECON 10.00
&END NOSE
&END THERMOSTAT
TEMPERATURE 303
&END MD
&TRAJECTORY
&EACH
MD 1
&END EACH
&END TRAJECTORY
&VELOCITIES OFF
&END VELOCITIES
&FORCES OFF
&END FORCES
&RESTART_HISTORY
&EACH
MD 500
&END EACH
&END RESTART_HISTORY
&RESTART
BACKUP_COPIES 3
&EACH
MD 1
&END EACH
&END RESTART
&END PRINT
&END
&EXT_RESTART
RESTART_FILE_NAME nvt-1.restart
&END

Marcella Iannuzzi
Nov 25, 2022, 4:15:43 AM11/25/22
to cp2k
Dear Dmitrii
One has to find the conditions for which the noise term generates the correct average temperature, as measured by the equipartition theorem.
This leads to the correct sampling.
Gamma should be small, as long as the error in the forces is small. Under these conditions the dynamical properties are correct.
The first term of comparison to determine the accuracy of the SGCP description is obtained from the deviations in energy and forces with respect to the BO values at the same coordinates.
The force deviation has to have a vanishing average, and also that the distribution of errors should be Gaussian.
Regards
Marcella

Moon Moon
Jan 11, 2023, 8:42:47 PM1/11/23
to cp2k
Dear Marcella
As you said, the accuracy of the SGCP description should be determined respect to the BO value at the same coordinates.
So do you mean that the BOMD should be set with ENSEMBLE LANGEVIN and converge per SCF step ?
Moon
Marcella Iannuzzi
Jan 12, 2023, 4:25:42 AM1/12/23
to cp2k
Dear Moon,
no, I mean that one should take the coordinates generated by SGCP and recalculate energies and forces by requesting a tight convergence of the SCF,
which means, retrieving the BO electronic structure and forces for the exact same coordinates.
These BO energy and forces can be compared to the energy and forces computed along the SGCP, which in turn should have been saved during the SGCP run.
From the average and distribution of the deviations one can evaluate the accuracy of the SGCP run.
Regards
Marcella
Moon Moon
Jan 12, 2023, 9:18:10 PM1/12/23
to cp2k
Dear Iannuzzi
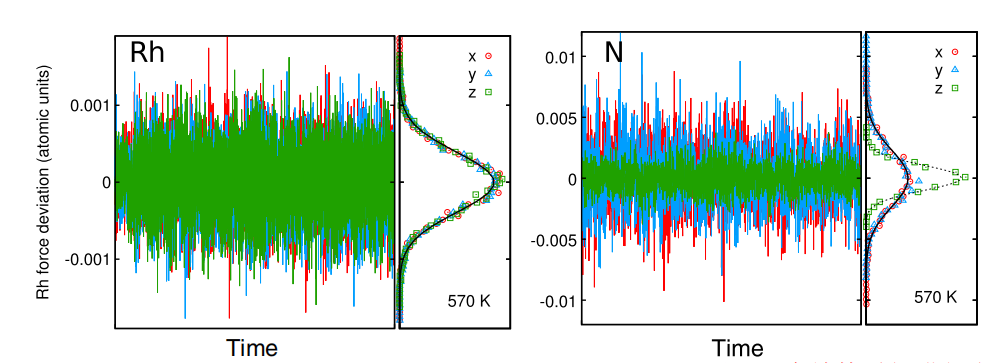
Many thanks for your detailed explanation.
I have realized how to evaluate the accuracy of SGCPMD.
One more question, in your paper "Second generation Car-Parrinello MD: application to the h-BN/Rh(111) nanomesh",
you wrote that " Configurations have been extracted every 12 fs from a trajectory of 180 fs" in the note for Fig. 3.
So there should be 15 data corresponding to single point energy calculation of 15 configurations.
Why are there so many data in Fig. 3 ?
Reply all
Reply to author
Forward
0 new messages