drift of center of mass

Wei Lai
Dear cp2k users and developers,
I am testing a NVE simulation with velocity scaling for crystalline CaO (300 K) and having questions regarding the drift of center of mass.
In a regular NVE simulation, I am expecting that (1) the simulation box is fixed and positions of atoms can be in fractional/scaled or Cartesian units, (2) if atoms moved out of the simulation box, they can be folded back to the box or left unchanged, (3) the mass-averaged position centers oscillate around x, y, z direction. I am showing the center of mass positions from VASP, which are consistent with the expectation.
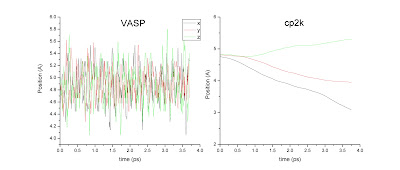
If I do the same for cp2k (input file enclosed), I am clearly seeing drift of center of mass. If I analyze the dynamics of the trajectory (now shown), it appears that both Ca and O atoms are diffusing instead of vibrating, as obtained from VASP trajectory.
I am wondering if you have seen similar behaviors or if I have the wrong settings in the input file.
Thanks, Wei

Matt W
Matt
Wei Lai

Ari Paavo Seitsonen
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+unsubscribe@googlegroups.com.
To post to this group, send email to cp...@googlegroups.com.
Visit this group at https://groups.google.com/group/cp2k.
For more options, visit https://groups.google.com/d/optout.
--
Ari Paavo Seitsonen / Ari.P.S...@iki.fi / http://www.iki.fi/~apsi/
Ecole Normale Supérieure (ENS), Département de Chimie, Paris
Mobile (F) : +33 789 37 24 25 (CH) : +41 79 71 90 935
Wei Lai
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To post to this group, send email to cp...@googlegroups.com.
Visit this group at https://groups.google.com/group/cp2k.
For more options, visit https://groups.google.com/d/optout.
Ari Paavo Seitsonen
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+unsubscribe@googlegroups.com.
To post to this group, send email to cp...@googlegroups.com.
Visit this group at https://groups.google.com/group/cp2k.
For more options, visit https://groups.google.com/d/optout.
Wei Lai
Ari Paavo Seitsonen
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+unsubscribe@googlegroups.com.
To post to this group, send email to cp...@googlegroups.com.
Visit this group at https://groups.google.com/group/cp2k.
For more options, visit https://groups.google.com/d/optout.