Guided AIMD reaction, CONSTRAINT, RESTRAINT, METADYNAMICS
521 views
Skip to first unread message

Dobromir A Kalchevski
Jan 19, 2023, 3:42:15 PM1/19/23
to cp2k
Hello everyone!
I want to make a molecule occasionally approach a surface and react with it in a NVT ensemble production run. I don't want it fixed in predetermined coordinates and I don't want it to fly away or to wait for too long of a trajectory.
For now I don't want to study the reaction's thermodynamics.
Experimentally the reaction occurs.
I tried the following approaches without success:
I defined a COLVAR for the distance between one surface atom (Si) and the center atom (C) of the gas phase reagent molecule (methane)
&COLVAR
&DISTANCE
AXIS XYZ !DEF XYZ, ANY COMBINATION
ATOMS 50 81
&END DISTANCE
&END COLVAR
&DISTANCE
AXIS XYZ !DEF XYZ, ANY COMBINATION
ATOMS 50 81
&END DISTANCE
&END COLVAR
and then I defined a CONSTRAINT
&CONSTRAINT
&COLLECTIVE
COLVAR 2
INTERMOLECULAR
TARGET [angstrom] 1.87
TARGET_GROWTH [angstrom*fs^-1] 0.25
TARGET_LIMIT [angstrom] 5.0
&END COLLECTIVE
&END CONSTRAINT
&COLLECTIVE
COLVAR 2
INTERMOLECULAR
TARGET [angstrom] 1.87
TARGET_GROWTH [angstrom*fs^-1] 0.25
TARGET_LIMIT [angstrom] 5.0
&END COLLECTIVE
&END CONSTRAINT
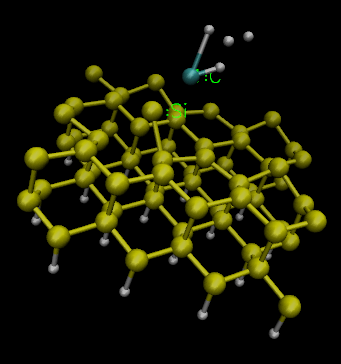
The result is that both atoms get closer too quickly and the system gets an unphysical geometry - the Si kind of moves too far away from it's lattice and the C abandons its hydrogens. The remperature rises too quickly to unbelievable degrees, the calculation slows down and eventually the program dies.
My cell is defined just fine, with enough space for the gas reagent to move around. I use a XYZ PBC with space left in one dimension so it effectively becomes 2D. In that space is the reacting molecule.
I tried playing with TARGET_GROWTH with many values from 0.01 to 5.0 and the result is always the same.
In this group I read that the moment TARGET is set the atoms are also set at that distance, so I tried switching TARGET and TARGET_LIMIT, but even then the result is the same - wrong geometry, they get too close too quickly even with very low TARGET_GROWTH, too high temperature - program dies.
- Is this a bug, because it makes no sense as expected behavior ?
I also tried using a RESTRAINT:
&CONSTRAINT
&COLLECTIVE
COLVAR 2
INTERMOLECULAR
TARGET [angstrom] 3.0
&RESTRAINT
K [kcalmol] 1.0
&END RESTRAINT
&END COLLECTIVE
&END CONSTRAINT
&COLLECTIVE
COLVAR 2
INTERMOLECULAR
TARGET [angstrom] 3.0
&RESTRAINT
K [kcalmol] 1.0
&END RESTRAINT
&END COLLECTIVE
&END CONSTRAINT
with the COLVAR being a plane between 3 surface atoms:
&COLVAR
&DISTANCE_POINT_PLANE
&POINT
TYPE GEO_CENTER
ATOMS 8
&END POINT
&POINT
TYPE GEO_CENTER
ATOMS 21
&END POINT
&POINT
TYPE GEO_CENTER
ATOMS 28
&END POINT
&POINT
TYPE GEO_CENTER
ATOMS 81
&END POINT
ATOMS_PLANE 1 2 3
ATOM_POINT 4
&END DISTANCE_POINT_PLANE
&END COLVAR
&DISTANCE_POINT_PLANE
&POINT
TYPE GEO_CENTER
ATOMS 8
&END POINT
&POINT
TYPE GEO_CENTER
ATOMS 21
&END POINT
&POINT
TYPE GEO_CENTER
ATOMS 28
&END POINT
&POINT
TYPE GEO_CENTER
ATOMS 81
&END POINT
ATOMS_PLANE 1 2 3
ATOM_POINT 4
&END DISTANCE_POINT_PLANE
&END COLVAR
and the gas phase reagent does not stop at the TARGET distance, instead it gets inserted into the Si lattice, pushing Si atoms away, inserting both C and H from the methane and one of the hydrogens flied far away. Temperature goes anomalously high, program slows down, dies. At least it took some time and steps to get to the wrong point.
- Why didn't the molecule stop at the surface, while experiencing the forces of the lattice atoms ?
- Why did it do beyond the TARGET distance ?
- Is this a bug ?
Can I use metadynamics in any way to push the molecule towards the surface for a reaction to occur ? Once again, I'm not interested in studying the FES or the rest of the thermodynamics.
I just need a way to get the gas phase reagent to occasionally get close enough for a reaction.
I think the answers to those question can be very useful for many.
I attach some pictures of the anomalous results. The first two are from the CONSTRAINT, the 3rd is from the RESTRAINT.
Best Regards,
Dobromir

{kind=link}
{kind=link}
{kind=link}
Marcella Iannuzzi
Jan 20, 2023, 7:21:37 AM1/20/23
to cp2k
Dear Dobromir
The TARGET_GROWTH is the rate of change of that target value, which in the example you posted is ridiculously high:
TARGET_GROWTH {Real}
Specifies the growth speed of the target value of the constrained collective variable.
The constraint with the value determined by the growth rate will be imposed, no matter which are the other forces in place.
At least until the calculation does not converge anymore and everything explodes.
Nothing of this is a sampling in the NVT ensemble, anyway.
If the goal is to sample the final state after the reaction has occurred, just start a simulation from the configuration of the reacted molecule.
If the goal is to find the reaction mechanism, there is no way out of studying the free energy surface or, in simpler cases, the potential energy surface.
An efficient way to find the potential minimal energy pathway in cases like yours is in general the nudged elastic band approach.
Regards
Marcella
Dobromir A Kalchevski
Jan 20, 2023, 7:35:31 AM1/20/23
to cp2k
Dear Marcella Iannuzzi,
Thank you for your reply.
Best Regards,
Dobromir
Dobromir A Kalchevski
Jan 20, 2023, 8:14:26 AM1/20/23
to cp2k
Still, my supervisor doesn't want me to study the FES, he simply wants me to make the reaction happen in dynamics. He wants the molecules to move around and eventually a reaction to occur.
Even if I try
&CONSTRAINT
&COLLECTIVE
COLVAR 2
INTERMOLECULAR
&COLLECTIVE
COLVAR 2
INTERMOLECULAR
TARGET [angstrom] 5.0 !initial position in the xyz geometry file
TARGET_GROWTH [angstrom*fs^-1] 0.01
TARGET_LIMIT [angstrom] 2.0
&END COLLECTIVE
&END CONSTRAINT
TARGET_GROWTH [angstrom*fs^-1] 0.01
TARGET_LIMIT [angstrom] 2.0
&END COLLECTIVE
&END CONSTRAINT
cp2k drives the two atoms from the COLVAR to a distance of 2.0 angstrom IMMEDIATELY. Is this normal or is it a bug ?
And once again, why does RESTAINT with 1 kcal/mol K drive the molecule into the surface lattice, pushing lattice atoms around and integrating the C atom from the methane into the lattice ? Could it be due to DFTB parameterization or is it a bug or am I missing something ?
Best Regards,
Dobromir
Marcella Iannuzzi
Jan 20, 2023, 8:32:09 AM1/20/23
to cp2k
If there is an activation barrier in this reaction, this will never happen in the right way just crashing the molecule into the surface
Regards
Marcella
Dobromir A Kalchevski
Jan 20, 2023, 9:00:59 AM1/20/23
to cp2k
This is the colvar:
&COLVAR
&DISTANCE
AXIS XYZ !DEF XYZ, ANY COMBINATION
ATOMS 50 81
&END DISTANCE
&END COLVAR
&DISTANCE
AXIS XYZ !DEF XYZ, ANY COMBINATION
ATOMS 50 81
&END DISTANCE
&END COLVAR
and with restraint it moves the C atom from methane (reacting molecule) to a Si atom in the Si lattice, the way it's supposed to, it just goes too far.
Best Regards,
Dobromir
Dobromir A Kalchevski
Jan 20, 2023, 7:00:24 PM1/20/23
to cp2k
Dear Marcella Iannuzzi,
Thank you again for the clarifications on sampling final state, finding reaction mechanism and potential minimal energy pathway :D This was very useful.
Best Regards,
Dobromir
Reply all
Reply to author
Forward
0 new messages