On the large temperature fluctuation of proton water clusters running AIMD

Mengxu Li (Mengxu)
Dear all,
I' m simulating the AIMD of the H+(H2O)3 cluster. I have seen this work, Schran et al., J. Chem. Theory Comput., 16, 88 (2020).
The article explicitly mentioned used parameter settings when simulating AIMD in the article: the cutoff of 500Ry, TZV2P basis set, the time step of 0.25fs, temperature of 300K, Nosé chain thermostat, etc. We have used similar parameter settings and simulated 25ps, but the temperature fluctuation range is large and there is no trend of equilibrium .
Could you give me some advice? I would appreciate your assistance in my problem.
PS: the input and output files are attached.
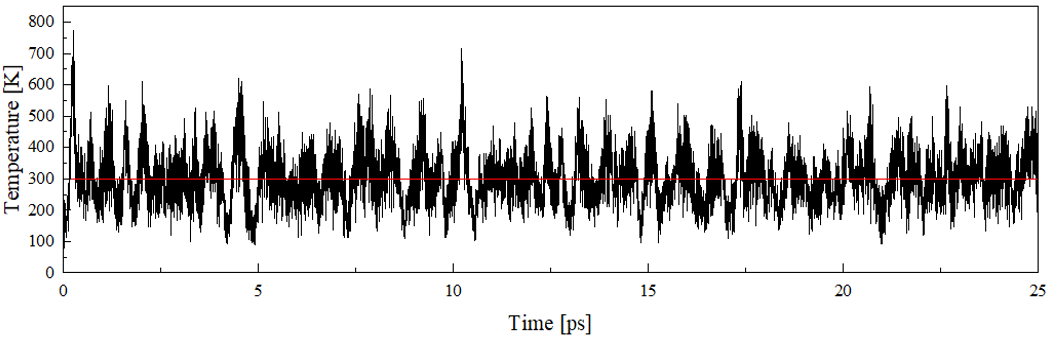
Yours sincerely,
LMX

钱洁
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/a338640d-10d7-4740-84f9-a68fdaedbf07n%40googlegroups.com.