Large discrepancy in xTB results from CP2K vs DFTB+
889 views
Skip to first unread message

Magnus Rahm
Sep 1, 2022, 6:48:35 AM9/1/22
to cp2k
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
Magnus Rahm
Sep 5, 2022, 2:40:58 AM9/5/22
to cp2k
For the record, the problem is the same in CP2K version 2022.1.

Jürg Hutter
Sep 5, 2022, 4:59:14 AM9/5/22
to cp...@googlegroups.com
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.

Anton Lytvynenko
Sep 5, 2022, 5:47:06 AM9/5/22
to cp...@googlegroups.com
Dear Magnus,
but do you have any reasons to expect that these methods must yield similar results? They are different and both are not so precise for the systems with complicated electronic effects.
Yours,
Anton
01.09.2022 12:48, Magnus Rahm пише:
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/2b7a001c-6e70-4e27-bb1d-cd6c1f4765f9n%40googlegroups.com.
Magnus Rahm
Sep 5, 2022, 5:58:31 AM9/5/22
to cp2k
Hi,
> Could you send a break down of the energies for the LiF molecule for
the two codes?
the two codes?
I just realized I used to small a cell in this case. I just reran the calculations for the LiF molecule and now CP2K and DFTB+ results match very well. Apologies for the confusion. This means I have only found the problem in periodic systems, which might provide a clue... Please let me know if you would like to see any other data.
> do you have any reasons to expect that these methods must
yield similar results? They are different and both are not so
precise for the systems with complicated electronic effects
My understanding is that they are the same method (GFN1-xTB) with at most small differences in default parameters, and I expected at least qualitatively the same results (on par with using say, PBE, with two different DFT codes). Maybe I've missed something important here?
Kind regards,
Magnus
Jürg Hutter
Sep 5, 2022, 6:07:03 AM9/5/22
to cp...@googlegroups.com
Hi
in any case it would be good to have a break down of energies for identical structures.
Known differences for the CP2K implementation:
1) There is no entropy term added by default. You can simulate that by using SMEARING
and diagonalization.
2) Dipole terms in the Coulomb energy are damped in the EWALD sum in order to avoid
problems with conditionally convergent energy contributions.
However, I assume the charges are the main problem.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
Sent: Monday, September 5, 2022 11:58 AM
To: cp2k
Subject: Re: [CP2K:17606] Large discrepancy in xTB results from CP2K vs DFTB+
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/2b7a001c-6e70-4e27-bb1d-cd6c1f4765f9n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/2b7a001c-6e70-4e27-bb1d-cd6c1f4765f9n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/4a80c6bd-6071-43fb-a996-b179d1767dc9n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/4a80c6bd-6071-43fb-a996-b179d1767dc9n%40googlegroups.com?utm_medium=email&utm_source=footer>.
in any case it would be good to have a break down of energies for identical structures.
Known differences for the CP2K implementation:
1) There is no entropy term added by default. You can simulate that by using SMEARING
and diagonalization.
2) Dipole terms in the Coulomb energy are damped in the EWALD sum in order to avoid
problems with conditionally convergent energy contributions.
However, I assume the charges are the main problem.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
To: cp2k
Subject: Re: [CP2K:17606] Large discrepancy in xTB results from CP2K vs DFTB+
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/4a80c6bd-6071-43fb-a996-b179d1767dc9n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/4a80c6bd-6071-43fb-a996-b179d1767dc9n%40googlegroups.com?utm_medium=email&utm_source=footer>.

Xavier Bidault
Sep 5, 2022, 6:18:26 AM9/5/22
to cp...@googlegroups.com
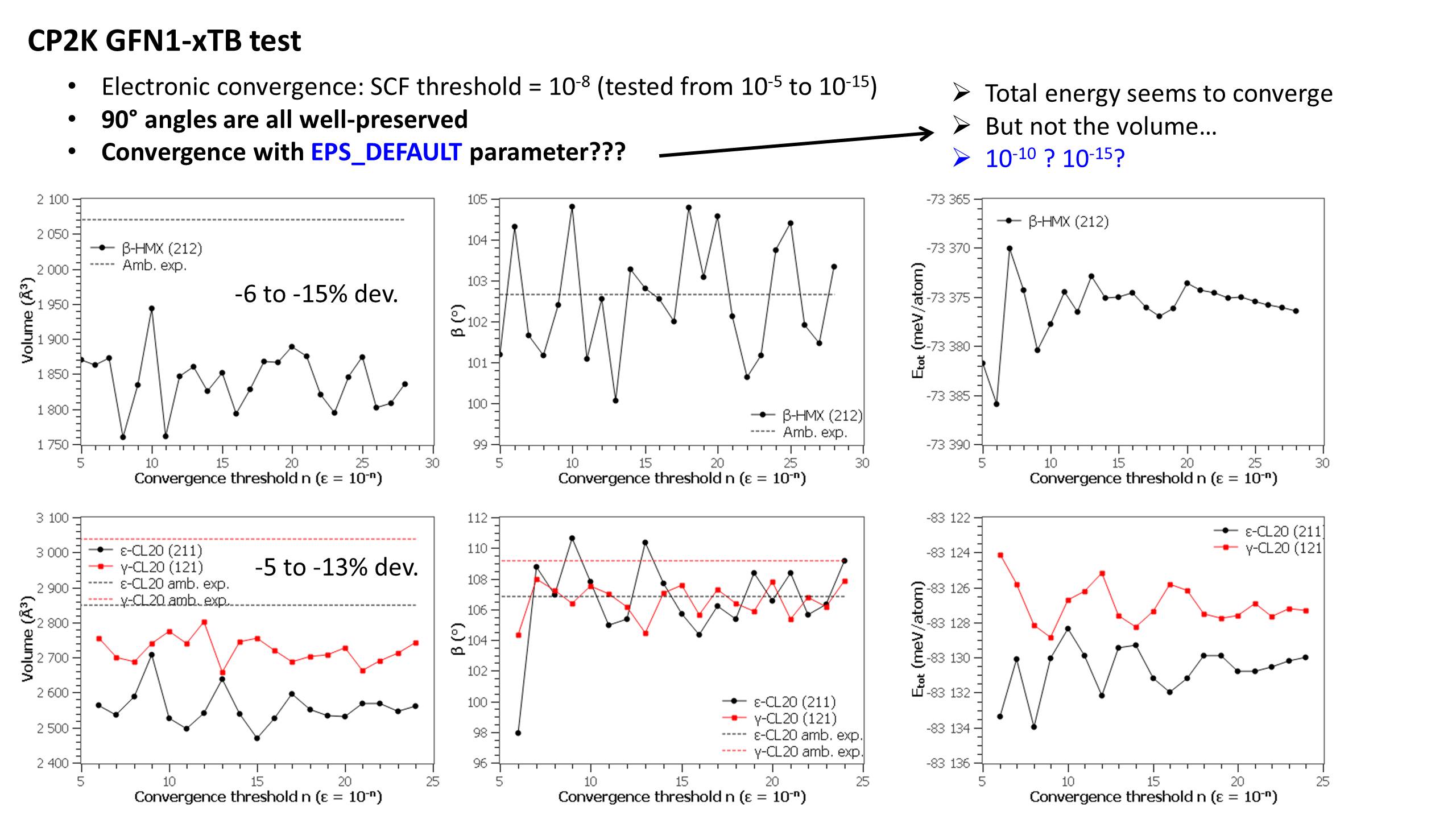
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075983A13AA78F53FFB422559F7F9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Magnus Rahm
Sep 5, 2022, 9:19:28 AM9/5/22
to cp2k
Hi,
Thank you for valuable input! Here's a breakdown of energies for a periodic LiO2 system (where CP2K and DFTB+ disagree).
CP2K:
Core Hamiltonian energy: -609.45757320827579
Repulsive potential energy: 2.86335541921533
Electronic energy: -65.73940900376786
DFTB3 3rd order energy: 9.00274299587460
Dispersion energy: -2.00065978643714
Correction for halogen bonding: 0.00000000000000
Total energy: -665.33154358339084
outer SCF iter = 1 RMS gradient = 0.49E-06 energy = -665.3315435834
outer SCF loop converged in 1 iterations or 10 steps
Repulsive potential energy: 2.86335541921533
Electronic energy: -65.73940900376786
DFTB3 3rd order energy: 9.00274299587460
Dispersion energy: -2.00065978643714
Correction for halogen bonding: 0.00000000000000
Total energy: -665.33154358339084
outer SCF iter = 1 RMS gradient = 0.49E-06 energy = -665.3315435834
outer SCF loop converged in 1 iterations or 10 steps
And the same system with DFTB+ (I don't know this is the best breakdown I can get from DFTB+? This info is from detailed.out.):
Fermi level: -0.1574062769 H -4.2832 eV
Band energy: -254.9890864567 H -6938.6061 eV
TS: 0.0000000000 H 0.0000 eV
Band free energy (E-TS): -254.9890864567 H -6938.6061 eV
Extrapolated E(0K): -254.9890864567 H -6938.6061 eV
Input / Output electrons (q): 864.0000000000 864.0000000000
Energy H0: -610.3586854777 H -16608.7049 eV
Energy SCC: 13.1915555608 H 358.9605 eV
Total Electronic energy: -597.1671299169 H -16249.7444 eV
Repulsive energy: 0.0000000000 H 0.0000 eV
Total energy: -597.1671299169 H -16249.7444 eV
Extrapolated to 0: -597.1671299169 H -16249.7444 eV
Total Mermin free energy: -597.1671299169 H -16249.7444 eV
Force related energy: -597.1671299169 H -16249.7444 eV
Band energy: -254.9890864567 H -6938.6061 eV
TS: 0.0000000000 H 0.0000 eV
Band free energy (E-TS): -254.9890864567 H -6938.6061 eV
Extrapolated E(0K): -254.9890864567 H -6938.6061 eV
Input / Output electrons (q): 864.0000000000 864.0000000000
Energy H0: -610.3586854777 H -16608.7049 eV
Energy SCC: 13.1915555608 H 358.9605 eV
Total Electronic energy: -597.1671299169 H -16249.7444 eV
Repulsive energy: 0.0000000000 H 0.0000 eV
Total energy: -597.1671299169 H -16249.7444 eV
Extrapolated to 0: -597.1671299169 H -16249.7444 eV
Total Mermin free energy: -597.1671299169 H -16249.7444 eV
Force related energy: -597.1671299169 H -16249.7444 eV
----------------------------------------------------------------------------------------------------------------
For reference, here are the equivalent breakdowns for the LiF molecule, where the total energies do match quite well.
CP2K:
Core Hamiltonian energy: -5.57594122418510
Repulsive potential energy: 0.00036401843654
Electronic energy: 0.08477836575096
DFTB3 3rd order energy: -0.00385103760005
Dispersion energy: -0.00008325087778
Correction for halogen bonding: 0.00000000000000
Total energy: -5.49473312847544
outer SCF iter = 1 RMS gradient = 0.12E-06 energy = -5.4947331285
outer SCF loop converged in 1 iterations or 25 steps
DFTB+
Fermi level: -0.3434874008 H -9.3468 eV
Band energy: -3.7493389034 H -102.0247 eV
TS: 0.0000000000 H 0.0000 eV
Band free energy (E-TS): -3.7493389034 H -102.0247 eV
Extrapolated E(0K): -3.7493389034 H -102.0247 eV
Input / Output electrons (q): 8.0000000444 8.0000000000
Energy H0: -5.5743451431 H -151.6856 eV
Energy SCC: 0.0807122067 H 2.1963 eV
Total Electronic energy: -5.4936329365 H -149.4894 eV
Repulsive energy: 0.0000000000 H 0.0000 eV
Total energy: -5.4936329365 H -149.4894 eV
Extrapolated to 0: -5.4936329365 H -149.4894 eV
Total Mermin free energy: -5.4936329365 H -149.4894 eV
Force related energy: -5.4936329365 H -149.4894 eV
Band energy: -3.7493389034 H -102.0247 eV
TS: 0.0000000000 H 0.0000 eV
Band free energy (E-TS): -3.7493389034 H -102.0247 eV
Extrapolated E(0K): -3.7493389034 H -102.0247 eV
Input / Output electrons (q): 8.0000000444 8.0000000000
Energy H0: -5.5743451431 H -151.6856 eV
Energy SCC: 0.0807122067 H 2.1963 eV
Total Electronic energy: -5.4936329365 H -149.4894 eV
Repulsive energy: 0.0000000000 H 0.0000 eV
Total energy: -5.4936329365 H -149.4894 eV
Extrapolated to 0: -5.4936329365 H -149.4894 eV
Total Mermin free energy: -5.4936329365 H -149.4894 eV
Force related energy: -5.4936329365 H -149.4894 eV
----------------------------------------------------------------------------------------------------------------
> I recently run variable-cell optimization of various molecular crystals
and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to
1e-24 (with EPS_SCF 1e-8), and no convergence happened.
Thank you for sharing this info! I tried a series of calculations with LiO2 using varying values of EPS_DEFAULT (using default EPS_SCF) and found the same effect; no convergence with EPS_DEFAULT (or perhaps unreasonably slow convergence). I attach a figure showing these results, including the energy broken down into the different parts as specified in the CP2K output. Note the energy scale, the changes with EPS_DEFAULT are really quite substantial. In the LiF (non-PBC) case, the corresponding curves look completely flat on the same scale. I don't know what to make of this result, but perhaps someone else does?
Magnus
Jürg Hutter
Sep 6, 2022, 5:37:21 AM9/6/22
to cp...@googlegroups.com
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
Sent: Monday, September 5, 2022 3:19 PM
To: cp2k
Subject: Re: [CP2K:17609] Re: Large discrepancy in xTB results from CP2K vs DFTB+
[X]
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com?utm_medium=email&utm_source=footer>.
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
To: cp2k
Subject: Re: [CP2K:17609] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Jürg Hutter
Sep 6, 2022, 11:36:30 AM9/6/22
to cp...@googlegroups.com
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Jürg Hutter <hut...@chem.uzh.ch>
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EEC38142F9D207742F549F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Jürg Hutter <hut...@chem.uzh.ch>
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Magnus Rahm
Sep 6, 2022, 4:29:17 PM9/6/22
to cp2k
Hi,
Thanks for the prompt fix! It made a significant difference but doesn't seem to resolve the discrepancy with DFTB+ (please see attached figure). Mulliken charges changed "in the right direction" but still differ very significantly compared to DFTB+.
Kind regards,
Magnus Rahm
Xavier Bidault
Sep 6, 2022, 8:16:23 PM9/6/22
to cp...@googlegroups.com
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759A3BAE12A4F8C0298072A9F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Magnus Rahm
Sep 7, 2022, 8:20:50 AM9/7/22
to cp2k
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
Xavier Bidault
Sep 7, 2022, 9:11:08 AM9/7/22
to cp...@googlegroups.com
>> Could you remind me how to update CP2K 2022.1 to include this bug fix?
> I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
I tried yesterday with git clone and started from scratch. The downloaded version (git:d56aa67) should have the xTB bug fix mentioned above (?), but when I run variable-cell optimizations of beta-HMX with various EPS_DEFAULT I find the exact same results as with CP2K 7.1. What's the right git version for this bug fix? Should I copy the git folder instead of using git clone? I mean, how to get the Trunk version you're talking about?
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com.
Magnus Rahm
Sep 7, 2022, 10:42:31 AM9/7/22
to cp2k
If you downloaded or cloned the repo at d56aa67 the bug fix is included. Safest approach to compare the versions is probably not a cell optimization but a single-point calculation (RUN_TYPE ENERGY_FORCE). The EV-curves in my post indicate that cell relaxation with the new version would (probably erroneously) have led to a very small cell as well, even though the underlying energies have changed significantly.
Kind regards,
Magnus Rahm
Xavier Bidault
Sep 7, 2022, 12:05:19 PM9/7/22
to cp...@googlegroups.com
So I won't consider this xTB issue fixed. Yet, I have no issue with EPS_DEFAULT when I use DFTB2 or DFTB3(diag) in CP2K for variable-cell optimization. Something is still off with xTB. But I must say I lack time to really look into it.
I also refer to this post (https://groups.google.com/g/cp2k/c/lESdjFfgq3g/m/drfONg10AQAJ). Actually I won't be looking for a straight comparison between CP2K and DFTB+, but rather for a steady/stable/reliable behavior of xTB@CP2K first (DFTB+ has other issues about the optimization algorithm, IMO). If of any help, please find attached my input script for beta-HMX using xTB@CP2K. Poisson/Ewald parameters are not enforced, but does CP2K set them up differently if EPS_DEFAULT is different? And why different EPS_DEFAULT would result in such large volume variations and unconverged behavior (see attached figure, not only for beta-HMX, and using supercells to have dimensions > 10 angstroms). The energy seems to converge for very low EPS_DEFAULT, but some spurious angle/volume variations remain. Similarly, I can show that xTB is not reliable for Molecular Dynamics, which prevents relevant prediction of the behavior under high pressure/temperature.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/e80f7efb-e11f-4e6c-bc4a-a20e0e234b57n%40googlegroups.com.
Jürg Hutter
Sep 16, 2022, 3:53:04 AM9/16/22
to cp...@googlegroups.com
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
outer SCF iter = 1 RMS gradient = 0.49E-06 energy = -665.3315435834<tel:(331)%20543-5834>
Fermi level: -0.3434874008<tel:(343)%20487-4008> H -9.3468 eV
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com?utm_medium=email&utm_source=footer>.
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
Sent: Wednesday, September 7, 2022 2:20 PM
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Magnus Rahm <mag...@compulartech.com>
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Band energy: -3.7493389034 H -102.0247 eV
TS: 0.0000000000 H 0.0000 eV
Band free energy (E-TS): -3.7493389034 H -102.0247 eV
Extrapolated E(0K): -3.7493389034 H -102.0247 eV
Input / Output electrons (q): 8.0000000444 8.0000000000
Energy H0: -5.5743451431<tel:(574)%20345-1431> H -151.6856 eV
TS: 0.0000000000 H 0.0000 eV
Band free energy (E-TS): -3.7493389034 H -102.0247 eV
Extrapolated E(0K): -3.7493389034 H -102.0247 eV
Input / Output electrons (q): 8.0000000444 8.0000000000
Xavier Bidault
Sep 18, 2022, 10:50:16 AM9/18/22
to cp...@googlegroups.com
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Xavier Bidault
Sep 18, 2022, 3:48:03 PM9/18/22
to cp...@googlegroups.com
Hi again,
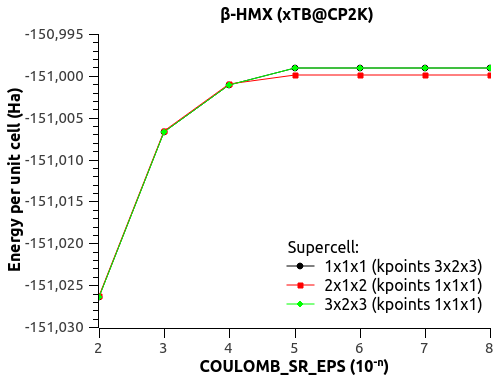
I found the documentation online for COULOMB_SR_EPS and COULOMB_SR_CUT. You'll find below more complete results of simulations using the last CP2K update (git:d529ce5) and variable-cell optimization of beta-HMX (unit cell and 3x2x3 k-points).
1) EPS_DEFAULT (1e-n below) dependency (with SCF 1e-8 and default COULOMB_SR_EPS 1e-3):
EPS_DEFAULT(1e-n) Energy(Ha) Volume(A3) beta(°)
6 -151.006625427635726 451.587706 101.776164
7 -151.006672807135971 451.590652 101.780407
8 -151.006676191174904 451.591670 101.780813
9 -151.006676832199673 451.591242 101.780877
10 -151.006676871358280 451.591222 101.780881
11 -151.006676887368030 451.591220 101.780880
6 -151.006625427635726 451.587706 101.776164
7 -151.006672807135971 451.590652 101.780407
8 -151.006676191174904 451.591670 101.780813
9 -151.006676832199673 451.591242 101.780877
10 -151.006676871358280 451.591222 101.780881
11 -151.006676887368030 451.591220 101.780880
-> Good convergence! No more weird variations. EPS_DEFAULT = 1e-8 is perfectly usable.
2) COULOMB_SR_EPS (1e-n below) dependency (with SCF 1e-8 and EPS_DEFAULT 1e-8):
COULOMB_SR_EPS(1e-n) Energy(Ha) Volume(A3) beta(°)
2 -151.026393509116332 466.813094 102.291412
3 -151.006676191174904 451.591670 101.780813
4 -151.001068739435084 467.465511 103.232087
5 -150.999031773201608 466.553038 103.882789
6 -150.999031773201438 466.553038 103.882789
7 -150.999031773201438 466.553038 103.882789
8 -150.999031773201438 466.553038 103.882789
9 -150.999031773201438 466.553038 103.882789
10 -150.999031773201438 466.553038 103.882789
2 -151.026393509116332 466.813094 102.291412
3 -151.006676191174904 451.591670 101.780813
4 -151.001068739435084 467.465511 103.232087
5 -150.999031773201608 466.553038 103.882789
6 -150.999031773201438 466.553038 103.882789
7 -150.999031773201438 466.553038 103.882789
8 -150.999031773201438 466.553038 103.882789
9 -150.999031773201438 466.553038 103.882789
10 -150.999031773201438 466.553038 103.882789
Actually, the default value or 1e-3 is the worst you could choose. I would recommend a default value for COULOMB_SR_EPS of 1e-5.
The behavior of xTB@CP2K is much more stable, and I would consider this issue solved. I just have to re-run a huge batch of simulations in the next 2 weeks with this update before submitting my paper ;-)
Thanks a lot!
Xavier
Jürg Hutter
Sep 19, 2022, 3:49:40 AM9/19/22
to cp...@googlegroups.com
Hi
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
Sent: Sunday, September 18, 2022 9:47 PM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17710] Re: Large discrepancy in xTB results from CP2K vs DFTB+
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com?utm_medium=email&utm_source=footer>.
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
Sent: Sunday, September 18, 2022 9:47 PM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17710] Re: Large discrepancy in xTB results from CP2K vs DFTB+
On Sun, Sep 18, 2022 at 9:50 AM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>> wrote:
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
On Fri, Sep 16, 2022 at 2:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com>>
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
Sent: Wednesday, September 7, 2022 2:20 PM
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
On Wednesday, September 7, 2022 at 2:16:23 AM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com> wrote:
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
On Tue, Sep 6, 2022 at 10:36 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com>
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com>>
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
On Monday, September 5, 2022 at 12:18:26 PM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com> wrote:
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
Le lun. 5 sept. 2022, 3:59 AM, Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> a écrit :
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com>>
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com>>.
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075983A13AA78F53FFB422559F7F9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EEC38142F9D207742F549F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759A3BAE12A4F8C0298072A9F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
Xavier Bidault
Sep 19, 2022, 9:39:40 PM9/19/22
to cp...@googlegroups.com
Hi Jürg,


I have a funny behavior with COULOMB_SR_EPS though, with the size of the system (replication of the unit cell to supercell). See the figures below.
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
Thanks,
Xavier
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759236DFA7DD6C1E25AA4B89F4D9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Xavier Bidault
Sep 19, 2022, 10:06:27 PM9/19/22
to cp...@googlegroups.com
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
Jürg Hutter
Sep 20, 2022, 6:53:42 AM9/20/22
to cp...@googlegroups.com
Hi
I can only guess here. The stress tensor might be a weak point of this type of spherical cutoff
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
To: cp...@googlegroups.com
Subject: Re: [CP2K:17714] Re: Large discrepancy in xTB results from CP2K vs DFTB+
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
Thanks,
Xavier
On Mon, Sep 19, 2022 at 2:49 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
Hi
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>>
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759236DFA7DD6C1E25AA4B89F4D9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com?utm_medium=email&utm_source=footer>.
I can only guess here. The stress tensor might be a weak point of this type of spherical cutoff
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
Sent: Tuesday, September 20, 2022 4:06 AM
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
To: cp...@googlegroups.com
Subject: Re: [CP2K:17714] Re: Large discrepancy in xTB results from CP2K vs DFTB+
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
On Mon, Sep 19, 2022 at 8:39 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>> wrote:
Hi Jürg,
Hi Jürg,
I have a funny behavior with COULOMB_SR_EPS though, with the size of the system (replication of the unit cell to supercell). See the figures below.
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
Thanks,
Xavier
On Mon, Sep 19, 2022 at 2:49 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
Hi
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
Sent: Sunday, September 18, 2022 9:47 PM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com>
On Sun, Sep 18, 2022 at 9:50 AM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>> wrote:
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
On Fri, Sep 16, 2022 at 2:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>> wrote:
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
Sent: Wednesday, September 7, 2022 2:20 PM
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
On Wednesday, September 7, 2022 at 2:16:23 AM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>> wrote:
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
On Tue, Sep 6, 2022 at 10:36 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>> wrote:
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
On Monday, September 5, 2022 at 12:18:26 PM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>> wrote:
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
Le lun. 5 sept. 2022, 3:59 AM, Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>> a écrit :
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>>.
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075983A13AA78F53FFB422559F7F9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EEC38142F9D207742F549F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759A3BAE12A4F8C0298072A9F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759236DFA7DD6C1E25AA4B89F4D9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
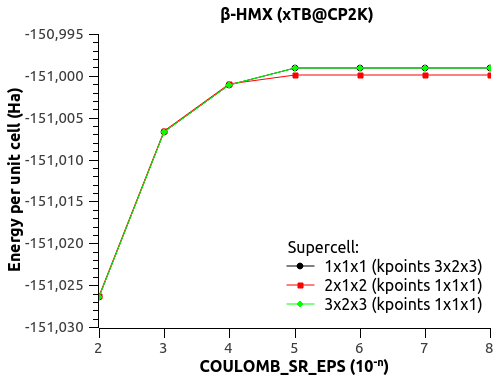{kind=link}
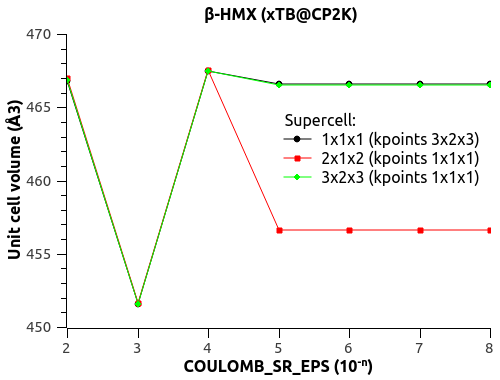{kind=link}
{kind=link}
Xavier Bidault
Sep 20, 2022, 9:16:53 PM9/20/22
to cp...@googlegroups.com
Hi Jürg,
I went further with the tests today, now varying COULOMB_SR_CUT (see pictures below). Actually, I like the idea of depending neither on COULOMB_SR_CUT nor on system size with COULOMB_SR_EPS = 1e-3, even though the resulting volume is 12.7% underestimated. But I would appreciate your insights on why it behaves like this (for COULOMB_SR_EPS = 1e-3). You talked earlier about per-element cutoffs. Is that it? And if so, are these per-element cutoffs parameters of xTB as published?
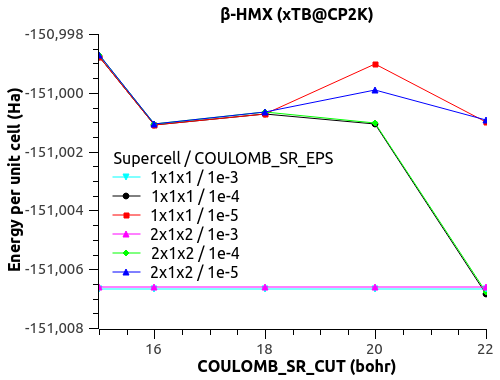
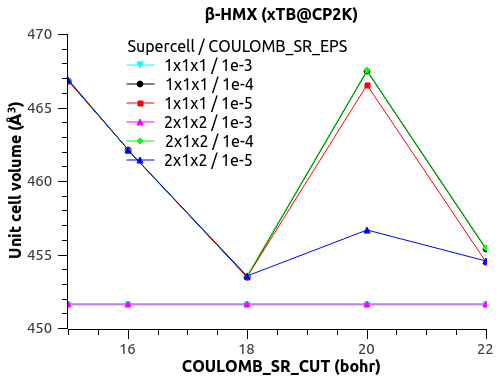
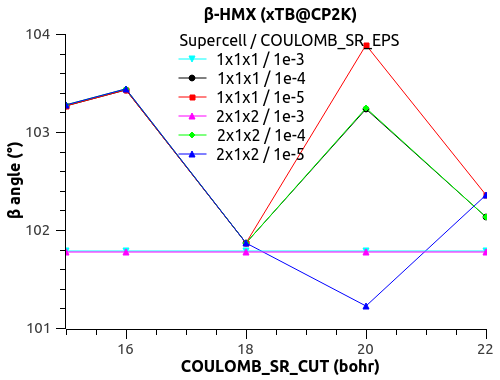
Thanks,
Xavier
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EDDA1E247702EA369A7A9F4C9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Xavier Bidault
Sep 24, 2022, 11:53:22 AM9/24/22
to cp...@googlegroups.com
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
Jürg Hutter
Sep 26, 2022, 3:45:16 AM9/26/22
to cp...@googlegroups.com
Hi
COULOMB_SR_EPS will result in an atom dependent cutoff. The atomic range is
dependent on the atomic hardness parameter in xTB.
However, I'm not satisfied with the current solution. It is much more stable than
the original (bug) one, but it still leads to arbitrary results when the parameters are
forced to the limits.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
Sent: Saturday, September 24, 2022 5:53 PM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17752] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EDDA1E247702EA369A7A9F4C9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWu55X%3DzztEAqbu-ep7EE%3D_4OgmMeBEc_Tw7UAd2A0F6w%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWu55X%3DzztEAqbu-ep7EE%3D_4OgmMeBEc_Tw7UAd2A0F6w%40mail.gmail.com?utm_medium=email&utm_source=footer>.
COULOMB_SR_EPS will result in an atom dependent cutoff. The atomic range is
dependent on the atomic hardness parameter in xTB.
However, I'm not satisfied with the current solution. It is much more stable than
the original (bug) one, but it still leads to arbitrary results when the parameters are
forced to the limits.
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
To: cp...@googlegroups.com
Subject: Re: [CP2K:17752] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
On Tue, Sep 20, 2022 at 8:16 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>> wrote:
Hi Jürg,
Hi Jürg,
I went further with the tests today, now varying COULOMB_SR_CUT (see pictures below). Actually, I like the idea of depending neither on COULOMB_SR_CUT nor on system size with COULOMB_SR_EPS = 1e-3, even though the resulting volume is 12.7% underestimated. But I would appreciate your insights on why it behaves like this (for COULOMB_SR_EPS = 1e-3). You talked earlier about per-element cutoffs. Is that it? And if so, are these per-element cutoffs parameters of xTB as published?
[image.png][image.png][image.png]
Thanks,
Xavier
Thanks,
Xavier
On Tue, Sep 20, 2022 at 5:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
Hi
I can only guess here. The stress tensor might be a weak point of this type of spherical cutoff
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
Hi
I can only guess here. The stress tensor might be a weak point of this type of spherical cutoff
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>>
Sent: Tuesday, September 20, 2022 4:06 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com>
Subject: Re: [CP2K:17714] Re: Large discrepancy in xTB results from CP2K vs DFTB+
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
On Mon, Sep 19, 2022 at 8:39 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>> wrote:
Hi Jürg,
Hi Jürg,
I have a funny behavior with COULOMB_SR_EPS though, with the size of the system (replication of the unit cell to supercell). See the figures below.
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
Thanks,
Xavier
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
Thanks,
Xavier
On Mon, Sep 19, 2022 at 2:49 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>> wrote:
Hi
Hi
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
Sent: Sunday, September 18, 2022 9:47 PM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>
On Sun, Sep 18, 2022 at 9:50 AM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>> wrote:
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
On Fri, Sep 16, 2022 at 2:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>> wrote:
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
Sent: Wednesday, September 7, 2022 2:20 PM
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
On Wednesday, September 7, 2022 at 2:16:23 AM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>> wrote:
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
On Tue, Sep 6, 2022 at 10:36 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>> wrote:
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> on behalf of Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
On Monday, September 5, 2022 at 12:18:26 PM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>> wrote:
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
Le lun. 5 sept. 2022, 3:59 AM, Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>> a écrit :
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>>>.
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075983A13AA78F53FFB422559F7F9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EEC38142F9D207742F549F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759A3BAE12A4F8C0298072A9F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759236DFA7DD6C1E25AA4B89F4D9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EDDA1E247702EA369A7A9F4C9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
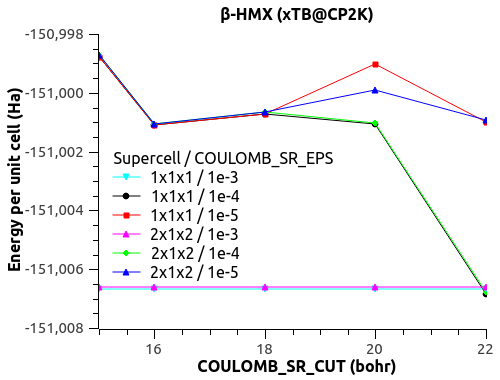{kind=link}
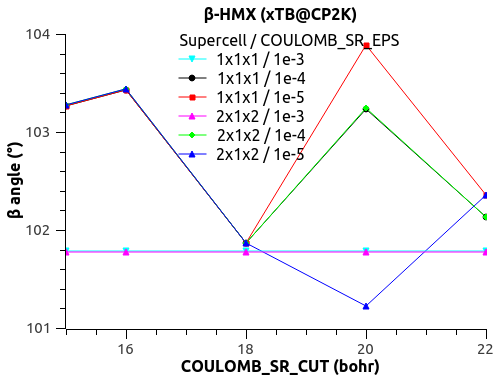{kind=link}
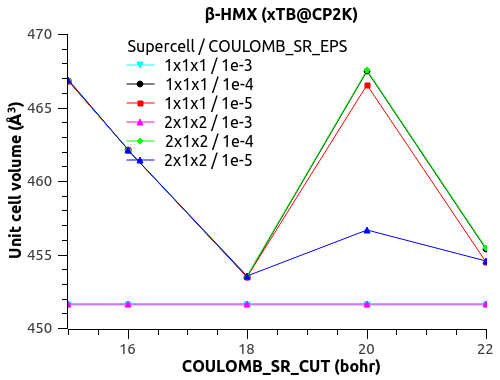{kind=link}
Xavier Bidault
Sep 28, 2022, 12:36:12 AM9/28/22
to cp...@googlegroups.com
Hi again Jürg,
For some reason, I would need the correspondence between the old and new adjustable parameters. I mean, with EPS_DEFAULT = 1e-10 in the previous CP2K version, what were the values of COULOMB_SR_EPS and COULOMB_SR_CUT in the same previous version?
Thank you,
Xavier
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759E9AA451C09F2F7FB258F9F529%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
Jürg Hutter
Sep 28, 2022, 3:44:25 AM9/28/22
to cp...@googlegroups.com
Hi
in the old version the cutoff was about 1.E-3 (calculated slightly different from now) and
there was no COULOMB_SR_CUT, but the maximum range was given by the extend of
the basis functions on an atom. This made the maximum range atom dependent and more
problematic dependent on EPS_DEFAULT.
You can still recover this (wrong) behavior with the keyword "OLD_COULOMB_DAMPING"
in the section "CP2K_INPUT / FORCE_EVAL / DFT / QS / XTB"
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
Sent: Wednesday, September 28, 2022 6:35 AM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17765] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi again Jürg,
For some reason, I would need the correspondence between the old and new adjustable parameters. I mean, with EPS_DEFAULT = 1e-10 in the previous CP2K version, what were the values of COULOMB_SR_EPS and COULOMB_SR_CUT in the same previous version?
Thank you,
Xavier
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759E9AA451C09F2F7FB258F9F529%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVrK-9mb6iJ0HAq7WHWaabPQpqMQho68Xh9LjRCG-jF7A%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVrK-9mb6iJ0HAq7WHWaabPQpqMQho68Xh9LjRCG-jF7A%40mail.gmail.com?utm_medium=email&utm_source=footer>.
in the old version the cutoff was about 1.E-3 (calculated slightly different from now) and
there was no COULOMB_SR_CUT, but the maximum range was given by the extend of
the basis functions on an atom. This made the maximum range atom dependent and more
problematic dependent on EPS_DEFAULT.
You can still recover this (wrong) behavior with the keyword "OLD_COULOMB_DAMPING"
in the section "CP2K_INPUT / FORCE_EVAL / DFT / QS / XTB"
regards
JH
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
To: cp...@googlegroups.com
Subject: Re: [CP2K:17765] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi again Jürg,
For some reason, I would need the correspondence between the old and new adjustable parameters. I mean, with EPS_DEFAULT = 1e-10 in the previous CP2K version, what were the values of COULOMB_SR_EPS and COULOMB_SR_CUT in the same previous version?
Thank you,
Xavier
On Mon, Sep 26, 2022 at 2:45 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
Hi
COULOMB_SR_EPS will result in an atom dependent cutoff. The atomic range is
dependent on the atomic hardness parameter in xTB.
However, I'm not satisfied with the current solution. It is much more stable than
the original (bug) one, but it still leads to arbitrary results when the parameters are
forced to the limits.
regards
JH
________________________________________
Hi
COULOMB_SR_EPS will result in an atom dependent cutoff. The atomic range is
dependent on the atomic hardness parameter in xTB.
However, I'm not satisfied with the current solution. It is much more stable than
the original (bug) one, but it still leads to arbitrary results when the parameters are
forced to the limits.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>>
Sent: Saturday, September 24, 2022 5:53 PM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com>
Subject: Re: [CP2K:17752] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
On Tue, Sep 20, 2022 at 8:16 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>> wrote:
Hi Jürg,
Hi Jürg,
I went further with the tests today, now varying COULOMB_SR_CUT (see pictures below). Actually, I like the idea of depending neither on COULOMB_SR_CUT nor on system size with COULOMB_SR_EPS = 1e-3, even though the resulting volume is 12.7% underestimated. But I would appreciate your insights on why it behaves like this (for COULOMB_SR_EPS = 1e-3). You talked earlier about per-element cutoffs. Is that it? And if so, are these per-element cutoffs parameters of xTB as published?
[image.png][image.png][image.png]
Thanks,
Xavier
[image.png][image.png][image.png]
Thanks,
Xavier
On Tue, Sep 20, 2022 at 5:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>> wrote:
Hi
Hi
I can only guess here. The stress tensor might be a weak point of this type of spherical cutoff
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>
Sent: Tuesday, September 20, 2022 4:06 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>
Subject: Re: [CP2K:17714] Re: Large discrepancy in xTB results from CP2K vs DFTB+
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
On Mon, Sep 19, 2022 at 8:39 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>> wrote:
Hi Jürg,
Hi Jürg,
I have a funny behavior with COULOMB_SR_EPS though, with the size of the system (replication of the unit cell to supercell). See the figures below.
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
Thanks,
Xavier
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
Thanks,
Xavier
On Mon, Sep 19, 2022 at 2:49 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>> wrote:
Hi
Hi
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
Sent: Sunday, September 18, 2022 9:47 PM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>
On Sun, Sep 18, 2022 at 9:50 AM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>> wrote:
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
On Fri, Sep 16, 2022 at 2:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>> wrote:
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>>
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
Sent: Wednesday, September 7, 2022 2:20 PM
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
On Wednesday, September 7, 2022 at 2:16:23 AM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>> wrote:
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
On Tue, Sep 6, 2022 at 10:36 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>> wrote:
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> on behalf of Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>>
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>>
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
On Monday, September 5, 2022 at 12:18:26 PM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>> wrote:
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
Le lun. 5 sept. 2022, 3:59 AM, Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>> a écrit :
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>>
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>>>.
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075983A13AA78F53FFB422559F7F9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EEC38142F9D207742F549F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759A3BAE12A4F8C0298072A9F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759236DFA7DD6C1E25AA4B89F4D9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EDDA1E247702EA369A7A9F4C9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWu55X%3DzztEAqbu-ep7EE%3D_4OgmMeBEc_Tw7UAd2A0F6w%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWu55X%3DzztEAqbu-ep7EE%3D_4OgmMeBEc_Tw7UAd2A0F6w%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759E9AA451C09F2F7FB258F9F529%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
Xavier Bidault
Sep 29, 2022, 9:26:44 AM9/29/22
to cp...@googlegroups.com
Hi Jürg,
I agree with the problematic behavior of the old version, but I had good results with EPS_DEFAULT = 1e-10 that I don't have anymore with the new version, whatever the parameters. I tried COULOMB_SR_EPS from 1e-2 to 1e-5 and COULOMB_SR_CUT from 15 to 22 bohr. That's why I was asking for the equivalent parameters in the new version (equivalent to EPS_DEFAULT = 1e-10). The default
SlaterRadialThresholdin AMS suite is 1e-5. Is this number related to COULOMB_SR_EPS in CP2K?
As a side, how long do you think it can take to implement the D2 and UFF vdw modules for xTB? Since UFF vdw is available for DFTB, it may be straightforward? And the D2 and D3(BJ) modules for DFTB as well? You may have seen the papers showing that DFTB + D3 or D3(BJ) yields very overestimated density for liquid water, even in ambient conditions with MD, which can be restored by using UFF vdw, despite less accurate. I would love the ability to cross the options.
Let me know!
Thanks,
Xavier
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB0753DC2857017758F9C385989F549%40ZRAP278MB0753.CHEP278.PROD.OUTLOOK.COM.
Jürg Hutter
Sep 29, 2022, 10:32:07 AM9/29/22
to cp...@googlegroups.com
Hi
the only way to recover the old behavior is to use OLD_COULOMB_DAMPING. The new
parameters are then ignored. There is no setting of COULOMB_SR_EPS and COULOMB_SR_CUT
that would emulate the old implementation.
Yes, having other vdW models working with DFTB/xTB is rather straightforward. However, I'm
hesitant (especially for xTB) to include this as it adds options to the input and risks to
be misused or causing confusion.
regards
Juerg
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
Sent: Thursday, September 29, 2022 3:26 PM
To: cp...@googlegroups.com
Subject: Re: [CP2K:17776] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi Jürg,
I agree with the problematic behavior of the old version, but I had good results with EPS_DEFAULT = 1e-10 that I don't have anymore with the new version, whatever the parameters. I tried COULOMB_SR_EPS from 1e-2 to 1e-5 and COULOMB_SR_CUT from 15 to 22 bohr. That's why I was asking for the equivalent parameters in the new version (equivalent to EPS_DEFAULT = 1e-10). The default
SlaterRadialThreshold in AMS suite is 1e-5. Is this number related to COULOMB_SR_EPS in CP2K?
As a side, how long do you think it can take to implement the D2 and UFF vdw modules for xTB? Since UFF vdw is available for DFTB, it may be straightforward? And the D2 and D3(BJ) modules for DFTB as well? You may have seen the papers showing that DFTB + D3 or D3(BJ) yields very overestimated density for liquid water, even in ambient conditions with MD, which can be restored by using UFF vdw, despite less accurate. I would love the ability to cross the options.
Let me know!
Thanks,
Xavier
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB0753DC2857017758F9C385989F549%40ZRAP278MB0753.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWXo3mpKRBgwBz6Z%3DAY1eagqr%3Dat_MonF9BwRd17u%2B7%2BQ%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWXo3mpKRBgwBz6Z%3DAY1eagqr%3Dat_MonF9BwRd17u%2B7%2BQ%40mail.gmail.com?utm_medium=email&utm_source=footer>.
the only way to recover the old behavior is to use OLD_COULOMB_DAMPING. The new
parameters are then ignored. There is no setting of COULOMB_SR_EPS and COULOMB_SR_CUT
that would emulate the old implementation.
Yes, having other vdW models working with DFTB/xTB is rather straightforward. However, I'm
hesitant (especially for xTB) to include this as it adds options to the input and risks to
be misused or causing confusion.
regards
Juerg
________________________________________
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Xavier Bidault <jazz...@gmail.com>
To: cp...@googlegroups.com
Subject: Re: [CP2K:17776] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi Jürg,
I agree with the problematic behavior of the old version, but I had good results with EPS_DEFAULT = 1e-10 that I don't have anymore with the new version, whatever the parameters. I tried COULOMB_SR_EPS from 1e-2 to 1e-5 and COULOMB_SR_CUT from 15 to 22 bohr. That's why I was asking for the equivalent parameters in the new version (equivalent to EPS_DEFAULT = 1e-10). The default
SlaterRadialThreshold in AMS suite is 1e-5. Is this number related to COULOMB_SR_EPS in CP2K?
As a side, how long do you think it can take to implement the D2 and UFF vdw modules for xTB? Since UFF vdw is available for DFTB, it may be straightforward? And the D2 and D3(BJ) modules for DFTB as well? You may have seen the papers showing that DFTB + D3 or D3(BJ) yields very overestimated density for liquid water, even in ambient conditions with MD, which can be restored by using UFF vdw, despite less accurate. I would love the ability to cross the options.
Let me know!
Thanks,
Xavier
On Wed, Sep 28, 2022 at 2:44 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>> wrote:
Hi
in the old version the cutoff was about 1.E-3 (calculated slightly different from now) and
there was no COULOMB_SR_CUT, but the maximum range was given by the extend of
the basis functions on an atom. This made the maximum range atom dependent and more
problematic dependent on EPS_DEFAULT.
You can still recover this (wrong) behavior with the keyword "OLD_COULOMB_DAMPING"
in the section "CP2K_INPUT / FORCE_EVAL / DFT / QS / XTB"
regards
JH
________________________________________
Hi
in the old version the cutoff was about 1.E-3 (calculated slightly different from now) and
there was no COULOMB_SR_CUT, but the maximum range was given by the extend of
the basis functions on an atom. This made the maximum range atom dependent and more
problematic dependent on EPS_DEFAULT.
You can still recover this (wrong) behavior with the keyword "OLD_COULOMB_DAMPING"
in the section "CP2K_INPUT / FORCE_EVAL / DFT / QS / XTB"
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com> <cp...@googlegroups.com<mailto:cp...@googlegroups.com>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com>>
Sent: Wednesday, September 28, 2022 6:35 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com>
Subject: Re: [CP2K:17765] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi again Jürg,
For some reason, I would need the correspondence between the old and new adjustable parameters. I mean, with EPS_DEFAULT = 1e-10 in the previous CP2K version, what were the values of COULOMB_SR_EPS and COULOMB_SR_CUT in the same previous version?
Thank you,
Xavier
Hi again Jürg,
For some reason, I would need the correspondence between the old and new adjustable parameters. I mean, with EPS_DEFAULT = 1e-10 in the previous CP2K version, what were the values of COULOMB_SR_EPS and COULOMB_SR_CUT in the same previous version?
Thank you,
Xavier
On Mon, Sep 26, 2022 at 2:45 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>> wrote:
Hi
Hi
COULOMB_SR_EPS will result in an atom dependent cutoff. The atomic range is
dependent on the atomic hardness parameter in xTB.
However, I'm not satisfied with the current solution. It is much more stable than
the original (bug) one, but it still leads to arbitrary results when the parameters are
forced to the limits.
regards
JH
________________________________________
dependent on the atomic hardness parameter in xTB.
However, I'm not satisfied with the current solution. It is much more stable than
the original (bug) one, but it still leads to arbitrary results when the parameters are
forced to the limits.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>
Sent: Saturday, September 24, 2022 5:53 PM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>
Subject: Re: [CP2K:17752] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
Hi Jürg,
I would appreciate your insights about the per-element cutoffs. See my previous email. Is it a feature of xTB or CP2K?
Thank you,
Xavier
On Tue, Sep 20, 2022 at 8:16 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>> wrote:
Hi Jürg,
Hi Jürg,
I went further with the tests today, now varying COULOMB_SR_CUT (see pictures below). Actually, I like the idea of depending neither on COULOMB_SR_CUT nor on system size with COULOMB_SR_EPS = 1e-3, even though the resulting volume is 12.7% underestimated. But I would appreciate your insights on why it behaves like this (for COULOMB_SR_EPS = 1e-3). You talked earlier about per-element cutoffs. Is that it? And if so, are these per-element cutoffs parameters of xTB as published?
[image.png][image.png][image.png]
Thanks,
Xavier
[image.png][image.png][image.png]
Thanks,
Xavier
On Tue, Sep 20, 2022 at 5:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>> wrote:
Hi
Hi
I can only guess here. The stress tensor might be a weak point of this type of spherical cutoff
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
implementation of long-ranged forces. Subtle changes of symmetry (size of your computational box,
k-points) together with the cutoff radius might cause changes in the stress tensor.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>
Sent: Tuesday, September 20, 2022 4:06 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>
Subject: Re: [CP2K:17714] Re: Large discrepancy in xTB results from CP2K vs DFTB+
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
I just tried with GMAX = 75 or 125 for replication 2x1x2 and COULOMB_SR_EPS = 1e-5, same result as above (lower energy than the unit cell and 3x2x3 supercell, lower volume and lower beta angle).
So the problem may not be the Ewald part.
Is there any known issue with small COULOMB_SR_EPS?
On Mon, Sep 19, 2022 at 8:39 PM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>> wrote:
Hi Jürg,
Hi Jürg,
I have a funny behavior with COULOMB_SR_EPS though, with the size of the system (replication of the unit cell to supercell). See the figures below.
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
Thanks,
Xavier
For COULOMB_SR_EPS = 1e-2 to 1e-4, there is no variation with the system size, which is good. (Naively?)
For COULOMB_SR_EPS >= 1e-5, there is a deviation, but only for the replication 2x1x2 of the supercell. I have checked with denser k-points 2x2x2 but the behavior is the same.
Would that mean that COULOMB_SR_EPS = 1e-4 is the optimal value?
What could explain this behavior for small COULOMB_SR_EPS??
The automatic Ewald? The only difference is the G-space max. Miller index:
1x1x1 supercell -> 45 75 45
2x1x2 supercell -> 75 75 125
3x2x3 supercell -> 125 125 135
Could that be it?
Let me know what you think.
[image.png][image.png][image.png]
Thanks,
Xavier
On Mon, Sep 19, 2022 at 2:49 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>> wrote:
Hi
Hi
thank you for the quick tests. It seems to me that the small COULOMB_SR_EPS has the
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> on behalf of Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>>
effect that all cutoff values are determined by COULOMB_SR_CUT (20 bohr).
This is the reason all your results for 10^-5 and smaller are identical.
I will further investigate how to treat the 1/r^3 terms more efficiently, but this
will not have a high priority.
best regards
JH
________________________________________
Sent: Sunday, September 18, 2022 9:47 PM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>
On Sun, Sep 18, 2022 at 9:50 AM Xavier Bidault <jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>>> wrote:
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
Hi Jürg,
A quick test with EPS_DEFAULT of 1e-10 or 1e-11 yields practically the same variable-cell optimization now for bHMX. So that's better, even though I'll have to check it up with a larger panel of values and watch convergence.
What are the default values you chose for these parameters?
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
Are they dependent on the (automatic) Ewald parameters?
If I want to modify them, what would be the section in the input file?
Are they "per atom" or global parameters?
Thank you,
Xavier
On Fri, Sep 16, 2022 at 2:53 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>>> wrote:
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>>>
I have updated the Trunk version with a new patch for xTB. This should now have the
electrostatic energy calculated as originally expected. The long-range 1/r term is
handled by an Ewald sum (using SPME) and the remaining terms with an 1/r^3 contribution
are cut at an atom dependent distance. The strong dependence of this term on the
requested general accuracy (EPS_DEFAULT) should now be gone.
The range (*2) of this interaction is controlled by two keywords
COULOMB_SR_EPS : atom dependent range
COULOMB_SR_CUT : maximum range for all atoms
This neglects the long range character of the 1/r^3 terms that might affect especially the
stress tensor.
I hope this helps to stabilize simulations.
best regards
JH
________________________________________
Sent: Wednesday, September 7, 2022 2:20 PM
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
To: cp2k
Subject: Re: [CP2K:17622] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Btw, I can confirm that the energy now converges with EPS_DEFAULT also for the LiO2 system, although the convergence is perhaps a bit slower (=very small EPS_DEFAULT values needed) than what one might have expected (see LiO2-EPS_DEFAULT.pdf). The figure I attached in my previous post was made with EPS_DEFAULT at the default value, if I use 1e-24 I get a bit closer to DFTB+ but still there is a weird slope in the E-V curve (EV-LiO2.pdf).
Furthermore, I had a look at the energy broken down into its different contributions as a function of volume (LiO2-energies-split.pdf), and FWIW it indicates that the electronic energy is responsible for the unexpected slope in the E-V curve (perhaps that was already obvious?).
> Could you remind me how to update CP2K 2022.1 to include this bug fix?
I think the easiest approach is to use Docker (following these instructions: https://github.com/cp2k/cp2k/tree/master/tools/docker), unless you want to clone the CP2K repo from github and compile from scratch.
Kind regards,
Magnus Rahm
On Wednesday, September 7, 2022 at 2:16:23 AM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>> wrote:
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
Thank you. Could you remind me how to update CP2K 2022.1 to include this bug fix?
On Tue, Sep 6, 2022 at 10:36 AM Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>>> wrote:
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>>> on behalf of Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>>>
Hi
the updates are now on Github (Trunk version).
This should at least fix the strange behavior for changes of EPS_DEFAULT.
regards
JH
________________________________________
Sent: Tuesday, September 6, 2022 11:37 AM
To: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>>
Subject: Re: [CP2K:17614] Re: Large discrepancy in xTB results from CP2K vs DFTB+
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>>>
Hi
I think I found the problem. This is in fact a bug in CP2K and is related to the damping of
the "short range" part of the Coulomb term. As mentioned before this short range part
is not short range at all, even diverging in periodic systems. We use a damping function
for this term and the radius is taken from the range of the basis function on each atom.
The bug is now, that this range is not a constant but depends on EPS_DEFAULT.
I will work on a solution, but at least the default settings will cause considerable changes
in the energies of periodic systems.
regards
JH
________________________________________
On Monday, September 5, 2022 at 12:18:26 PM UTC+2 jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com><mailto:jazz...@gmail.com<mailto:jazz...@gmail.com>>>>> wrote:
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
I recently run variable-cell optimization of various molecular crystals and I found xTB@CP2K ultra sensitive to EPS_DEFAULT. Tested from 1e-5 to 1e-24 (with EPS_SCF 1e-8), and no convergence happened. I just ended up with EPS_DEFAULT 1e-10 as a "gut" choice. Also, the behavior of xTB@CP2K is doutfull with MD even at ambiant conditions, where the converged volume is barely larget than at 0K. Depending on EPS_DEFAULT, it can even be smaller at ambient T. Weird. The behavior of DFTB2@CP2K is far better.
I found that DFTB+ has other issues. xTB@DFTB+ has no convergence issue, but the recommended variable-cell optimization algorithm has flaws. The unit cell and a supercell does NOT always end up with related lattice parameters. The main issue is that some 90° angles are not preserved with DFTB+ whereas CP2K does (with no symmetry enforced, obviously). Some inconsistencies appears in DFTB+ with a lattice dimensions < 10 angstroms in the unit cell versus > 10 angstroms in the supercell. A proper tight mesh of k-points does not improve. So I'm afraid that xTB@DFTB+ (or DFTB+, actually) cannot be a relevant choice for crystal structure predictions, for instance.
xTB may be unreliable with CP2K and DFTB+, but for the different reasons above. You can check these weird behaviors with your own crystals of interest.
Xavier
Le lun. 5 sept. 2022, 3:59 AM, Jürg Hutter <hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch><mailto:hut...@chem.uzh.ch<mailto:hut...@chem.uzh.ch>>>>>> a écrit :
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
From: cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>> <cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com><mailto:cp...@googlegroups.com<mailto:cp...@googlegroups.com>>>>>> on behalf of Magnus Rahm <mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com><mailto:mag...@compulartech.com<mailto:mag...@compulartech.com>>>>>>
Hi
thank you for testing. Could you send a break down of the energies for the LiF molecule for
the two codes? That might help to recognize the source of the difference.
regards
JH
________________________________________
Sent: Monday, September 5, 2022 8:40 AM
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>>>.
To: cp2k
Subject: [CP2K:17599] Re: Large discrepancy in xTB results from CP2K vs DFTB+
For the record, the problem is the same in CP2K version 2022.1.
On Thursday, September 1, 2022 at 12:48:35 PM UTC+2 Magnus Rahm wrote:
Dear all,
I want to use CP2K (version 8.2, trying to get a more recent version compiled) together with xTB for a crystal containing Li and O. I get strange results already for a simple LiO2 crystal:
* There is a very large discrepancy compared to DFTB+ (version 22.1).
* Mulliken charges tend to be large, meaning that CHECK_ATOMIC_CHARGES stops the SCF. If I turn it off, the system tends to converge systematically to values just outside the "chemical range". Mulliken charges obtained by DFTB+ are significantly smaller (and within "chemical range").
* The energy-volume curve looks strange and very different from DFTB+.
I have tried converging with respect to system size and the EWALD / ALPHA and GMAX parameters, but they have only a marginal impact. I have tried similar calculations for a number of periodic systems. Sometimes I get agreement, sometimes not. I also tried calculations for CO and NO molecules which agree perfectly between CP2K and DFTB+, whereas an artificial LiF molecule does not.
A perhaps related issue was reported in https://groups.google.com/g/cp2k/c/oFwgGcQuySs but the solutions suggested there did not solve my problem.
I attach input scripts for CP2K and DFTB+, as well as a figure showing the E-V curve for LiO2 obtained with CP2K and DFTB+. I'm new to CP2K, DFTB+ and xTB so I suspect I have made some simple mistake, and any advice is appreciated.
Kind regards,
Magnus Rahm
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/c51e70a2-7f7a-4887-8ada-971d840a1b13n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075983A13AA78F53FFB422559F7F9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/7d455fce-a22e-44f1-ac48-18495f9553a5n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EEC38142F9D207742F549F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Buns...@googlegroups.com><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>>><mailto:cp2k%2Buns...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Buns...@googlegroups.com><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com>><mailto:cp2k%25252Buns...@googlegroups.com<mailto:cp2k%2525252Buns...@googlegroups.com><mailto:cp2k%2525252Buns...@googlegroups.com<mailto:cp2k%252525252Buns...@googlegroups.com>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759A3BAE12A4F8C0298072A9F7E9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com><mailto:cp2k%2525252Bu...@googlegroups.com<mailto:cp2k%252525252B...@googlegroups.com>>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com><mailto:cp2k%2525252Bu...@googlegroups.com<mailto:cp2k%252525252B...@googlegroups.com>>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com<https://groups.google.com/d/msgid/cp2k/1234148a-7a9a-4c6b-a0c9-13caabfe8811n%40googlegroups.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com><mailto:cp2k%2525252Bu...@googlegroups.com<mailto:cp2k%252525252B...@googlegroups.com>>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB075914FC480ED2BF2B9B7FEB9F489%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNU-0x-3VheOAWmo2woUsZ_kF1395vcq2zdAT6qnCjauow%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com><mailto:cp2k%25252Bun...@googlegroups.com<mailto:cp2k%2525252Bu...@googlegroups.com>>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759236DFA7DD6C1E25AA4B89F4D9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVivpKr8Pw-cEsRYhPYUdCU44UMUutMGT%3DvCmtBZ7P6pQ%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com><mailto:cp2k%252Buns...@googlegroups.com<mailto:cp2k%25252Bun...@googlegroups.com>>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759EDDA1E247702EA369A7A9F4C9%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWu55X%3DzztEAqbu-ep7EE%3D_4OgmMeBEc_Tw7UAd2A0F6w%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNWu55X%3DzztEAqbu-ep7EE%3D_4OgmMeBEc_Tw7UAd2A0F6w%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k%2Bunsu...@googlegroups.com<mailto:cp2k%252Buns...@googlegroups.com>>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZR0P278MB0759E9AA451C09F2F7FB258F9F529%40ZR0P278MB0759.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com><mailto:cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>>.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVrK-9mb6iJ0HAq7WHWaabPQpqMQho68Xh9LjRCG-jF7A%40mail.gmail.com<https://groups.google.com/d/msgid/cp2k/CAD0N%2BNVrK-9mb6iJ0HAq7WHWaabPQpqMQho68Xh9LjRCG-jF7A%40mail.gmail.com?utm_medium=email&utm_source=footer>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k%2Bunsu...@googlegroups.com>.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB0753DC2857017758F9C385989F549%40ZRAP278MB0753.CHEP278.PROD.OUTLOOK.COM.
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com<mailto:cp2k+uns...@googlegroups.com>.
Reply all
Reply to author
Forward
0 new messages