Doubt regarding CN analysis

Vaishali Chakraborty

Tali Mazor
I hope that helps,
--
You received this message because you are subscribed to the Google Groups "cBioPortal for Cancer Genomics Discussion Group" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cbioportal+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cbioportal/CAB5ADusOMn1i_Eboc520xmuZtKU1zepsM3tJ%3Dg3eqPr%3Dzu1TGQ%40mail.gmail.com.
Vaishali Chakraborty
Vaishali Chakraborty
Vaishali Chakraborty
Vaishali Chakraborty

Ritika Kundra
Vaishali Chakraborty
Vaishali Chakraborty

kun...@mskcc.org
Hi Vaishali,
Hmm I think you might need to consult with someone from CNA bioinformatics pipeline. But we did find a similar question on Biostar that might be helpful for you:
https://www.biostars.org/p/306180/
It might be actually helpful if you ask on Biostars.
Thanks,
Ritika
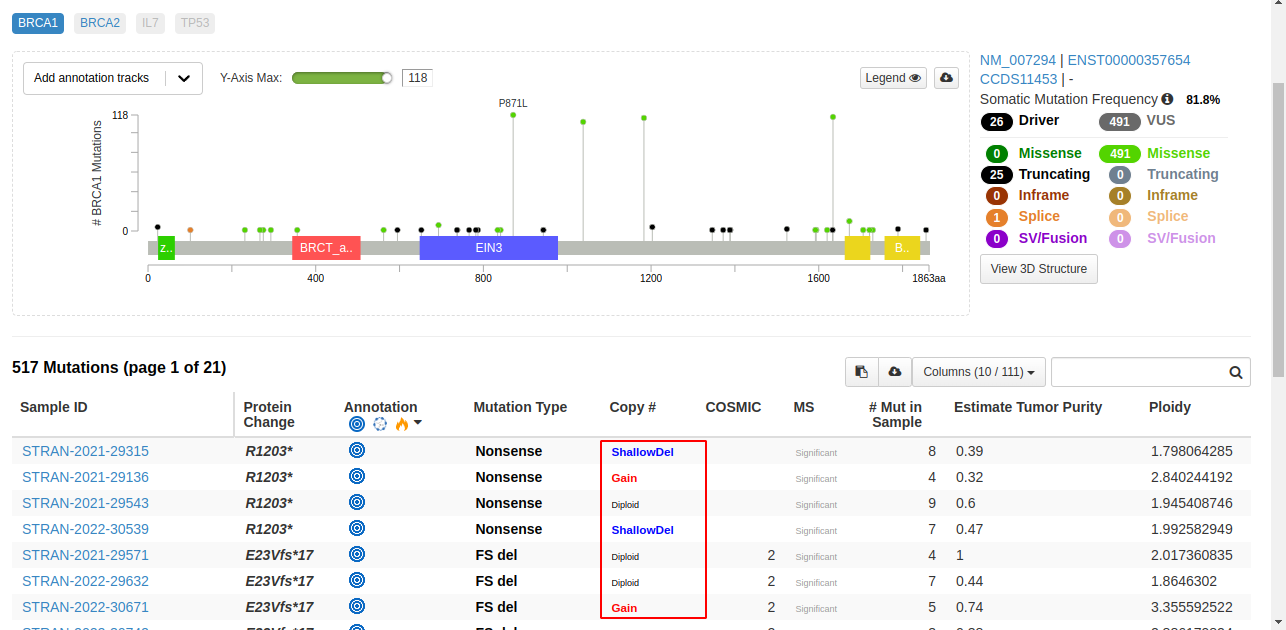
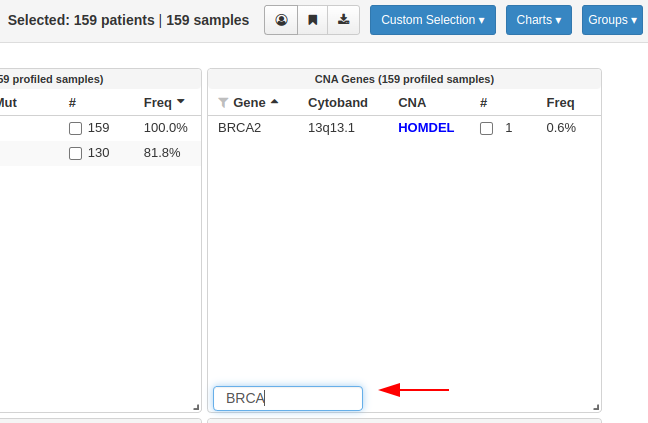
However, in the study view it shows only BRCA2 gene having 1 mutation, BRCA1 is completely missing and the numbers also look odd. In the above image itself I can see at least 4 samples with Copy# alterations. Please find the reference image below showing only the BRCA2 gene when BRCA is being searched. The profiled samples for mutations and copy number and all samples are same as all are tumor samples:
Looking forward to hearing from you.
Best regards,
Vaishali
--
You received this message because you are subscribed to the Google Groups "cBioPortal for Cancer Genomics Discussion Group" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cbioportal+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cbioportal/CAB5ADusOMn1i_Eboc520xmuZtKU1zepsM3tJ%3Dg3eqPr%3Dzu1TGQ%40mail.gmail.com.
=====================================================================
Please note that this e-mail and any files transmitted from
Memorial Sloan Kettering Cancer Center may be privileged, confidential,
and protected from disclosure under applicable law. If the reader of
this message is not the intended recipient, or an employee or agent
responsible for delivering this message to the intended recipient,
you are hereby notified that any reading, dissemination, distribution,
copying, or other use of this communication or any of its attachments
is strictly prohibited. If you have received this communication in
error, please notify the sender immediately by replying to this message
and deleting this message, any attachments, and all copies and backups
from your computer.
Vaishali Chakraborty
Vaishali Chakraborty
kun...@mskcc.org
Hi Vaishali,
There are many different methods used throughout MSK for CNA detection. One example is the MSK-IMPACT clinical series where they define their methods in these two publications:
https://pubmed.ncbi.nlm.nih.gov/25801821/
Error! Filename not specified.
However, in the study view it shows only BRCA2 gene having 1 mutation, BRCA1 is completely missing and the numbers also look odd. In the above image itself I can see at least 4 samples with Copy# alterations. Please find the reference image below showing only the BRCA2 gene when BRCA is being searched. The profiled samples for mutations and copy number and all samples are same as all are tumor samples:
Error! Filename not specified.