Evaluating Flexibility of 3 Different Proteins

Sherik726

Richard Gillilan
--
You received this message because you are subscribed to the Google Groups "BioXTAS RAW" group.
To unsubscribe from this group and stop receiving emails from it, send an email to bioxtas_raw...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/bioxtas_raw/1561abf4-2411-4cd3-99dc-dcbda041db5eo%40googlegroups.com.
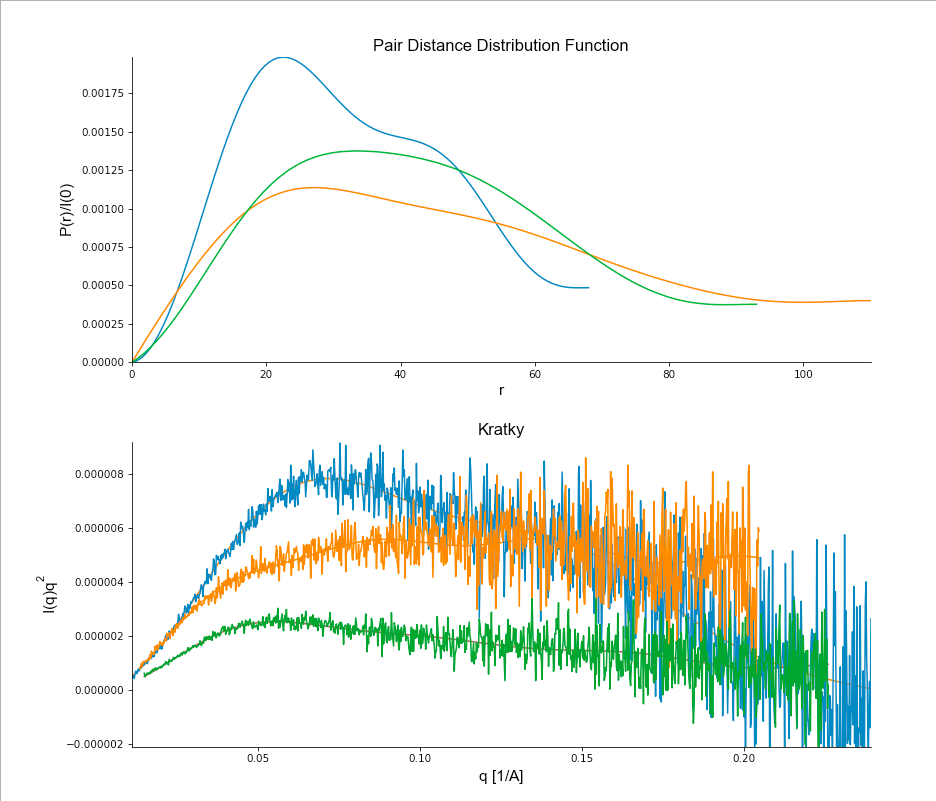
<Screen Shot 2020-07-30 at 11.04.29 AM.png>

Jesse Hopkins
To view this discussion on the web visit https://groups.google.com/d/msgid/bioxtas_raw/F4E571E6-870D-4568-BB36-6C42EC09F05A%40cornell.edu.

{kind=link}
kushol...@gmail.com
Two cents –
Check out https://pubmed.ncbi.nlm.nih.gov/21509745/
I frequently will use the Porod exponent as quantified in the program ScAtter and the flexibility plots described to characterize this property.
Also check out the work of Pau Bernado’s group characterizing polyubiquitin chains using SAXS and EOMs. Some great discussions of how flexibility manifests itself in SAS data and analysis at every stage.
Also see if your masses determined by Vc and I(0) greatly vary with those sensitive to protein volume and compactness like Vp and from the bead models. The latter calculations will be compromised by flexibility.
Kushol
To view this discussion on the web visit https://groups.google.com/d/msgid/bioxtas_raw/CAGRN2W1OZsk_sTAFzWFTUv45pvED89%3Dq8fK4jD51BUnmTNw0Ow%40mail.gmail.com.
Sherik726
Hello,
{kind=link}
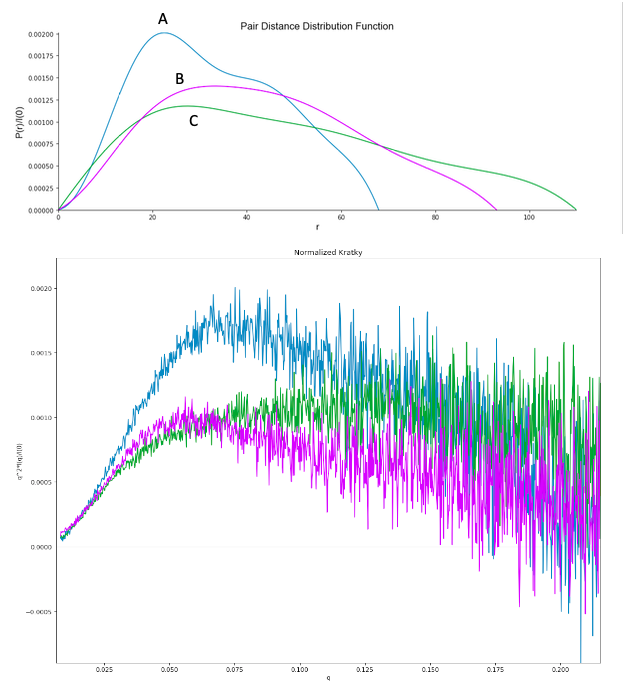{kind=link}
Sherik726
Richard
To unsubscribe from this group and stop receiving emails from it, send an email to bioxt...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/bioxtas_raw/1561abf4-2411-4cd3-99dc-dcbda041db5eo%40googlegroups.com.
<Screen Shot 2020-07-30 at 11.04.29 AM.png>
--
You received this message because you are subscribed to the Google Groups "BioXTAS RAW" group.
To unsubscribe from this group and stop receiving emails from it, send an email to bioxt...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/bioxtas_raw/F4E571E6-870D-4568-BB36-6C42EC09F05A%40cornell.edu.
--
You received this message because you are subscribed to the Google Groups "BioXTAS RAW" group.
To unsubscribe from this group and stop receiving emails from it, send an email to bioxt...@googlegroups.com.
Jesse Hopkins
--
You received this message because you are subscribed to the Google Groups "BioXTAS RAW" group.
To unsubscribe from this group and stop receiving emails from it, send an email to bioxtas_raw...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/bioxtas_raw/131d6e4f-d7bc-4812-8697-d721565ce539o%40googlegroups.com.