The error of cnvkit (file format)
238 views
Skip to first unread message

翰欽鄭
Feb 2, 2018, 4:20:03 AM2/2/18
to biovalidation
Hello:
I used bcbio to test my project, and I encountered a error below:
errorlog link
The error message said the file "T-LA-F03_S17_PE_cutadapt_QC-variant_regions.quantized-vrsubset-callableblocks.bed" is not the bed.
But I had check the file in work/bedprep/, the format and extension file name is bed file.
So I don't know what's wrong in this project.
Please help me to solve this problem.
Thank you.
PS: Below link is the config file
config file
I used bcbio to test my project, and I encountered a error below:
errorlog link
subprocess.CalledProcessError: Command 'set -o pipefail; export TMPDIR=/mnt/nfsfile/yourgene-academic-workspace-pool2/zhenghank/All_projects/OD20171203-CLA252/bcbio_test/OD20171203-merged/work/bcbiotx/tmpNCk_zf && /mnt/nfsfile/NAS6/zhenghank/bin/bcbio/anaconda/bin/cnvkit.py target /mnt/nfsfile/yourgene-academic-workspace-pool2/zhenghank/All_projects/OD20171203-CLA252/bcbio_test/OD20171203-merged/work/bedprep/T-LA-F03_S17_PE_cutadapt_QC-variant_regions.quantized-vrsubset-callableblocks.bed --split -o /mnt/nfsfile/yourgene-academic-workspace-pool2/zhenghank/All_projects/OD20171203-CLA252/bcbio_test/OD20171203-merged/work/bcbiotx/tmpNCk_zf/T-LA-F03_S17_PE_cutadapt_QC-target.bed --avg-size 250
Traceback (most recent call last):
File "/mnt/nfsfile/NAS6/zhenghank/bin/bcbio/anaconda/bin/cnvkit.py", line 13, in <module>
args.func(args)
File "/home/zhenghank/zhenghank/bin/bcbio/anaconda/lib/python2.7/site-packages/cnvlib/commands.py", line 229, in _cmd_target
regions = tabio.read_auto(args.interval)
File "/home/zhenghank/zhenghank/bin/bcbio/anaconda/lib/python2.7/site-packages/skgenome/tabio/__init__.py", line 108, in read_auto
fmt = sniff_region_format(infile)
File "/home/zhenghank/zhenghank/bin/bcbio/anaconda/lib/python2.7/site-packages/skgenome/tabio/__init__.py", line 253, in sniff_region_format
% (fname, ', '.join(format_patterns.keys()), line))
ValueError: File '/mnt/nfsfile/yourgene-academic-workspace-pool2/zhenghank/All_projects/OD20171203-CLA252/bcbio_test/OD20171203-merged/work/bedprep/T-LA-F03_S17_PE_cutadapt_QC-variant_regions.quantized-vrsubset-callableblocks.bed' does not appear to be a recognized format! (Any of: text, tab, interval, refflat, gff, bed)
First non-blank line:
NZ_CP022130.1 0 62322
' returned non-zero exit status 1
The error message said the file "T-LA-F03_S17_PE_cutadapt_QC-variant_regions.quantized-vrsubset-callableblocks.bed" is not the bed.
But I had check the file in work/bedprep/, the format and extension file name is bed file.
So I don't know what's wrong in this project.
Please help me to solve this problem.
Thank you.
PS: Below link is the config file
config file

Brad Chapman
Feb 2, 2018, 10:09:06 AM2/2/18
to 翰欽鄭, biovalidation
Thanks for the report and sorry about the issue. That's unexpected, I haven't
seen this problem before and when I look at the file manually or use the
skgenome.tabio.sniff_region_format test on the file you sent it correctly
detects it.
Some ideas to debug:
- Do you have the latest version of CNVkit?
$ bcbio_conda list | grep cnvkit
cnvkit 0.9.2a0 py27_2 bioconda
- Is it possible this is a filesystem issue accessing this file and re-running
will work cleanly?
Sorry to not know what is happening but hope these help some,
Brad
> [ text/plain ]
> Hello:
>
> I used bcbio to test my project, and I encountered a error below:
>
> errorlog link
> <https://gist.github.com/blueskys123/ebac0baa518692984ff053e72e91b98e>
>
> I used bcbio to test my project, and I encountered a error below:
>
> errorlog link
>
> --
> You received this message because you are subscribed to the Google Groups "biovalidation" group.
> To unsubscribe from this group and stop receiving emails from it, send an email to biovalidatio...@googlegroups.com.
> To post to this group, send email to bioval...@googlegroups.com.
> Visit this group at https://groups.google.com/group/biovalidation.
> To view this discussion on the web visit https://groups.google.com/d/msgid/biovalidation/7397f441-067d-4861-86a5-27dcd3f5a73b%40googlegroups.com.
> For more options, visit https://groups.google.com/d/optout.
> [ T-LA-F03_S17_PE_cutadapt_QC-variant_regions.quantized-vrsubset-callableblocks.bed: application/octet-stream ]
翰欽鄭
Feb 5, 2018, 12:45:12 AM2/5/18
to biovalidation
Thank you for your comment.
I check my cnvkit version, and I found my cnvkit version is py27_1, not py27_2.
Therefore, I updated my bcbio and reran the project.
The project works fine.
Thank you.
My cnvkit version:

Brad Chapman於 2018年2月2日星期五 UTC+8下午11時09分06秒寫道:
I check my cnvkit version, and I found my cnvkit version is py27_1, not py27_2.
Therefore, I updated my bcbio and reran the project.
The project works fine.
Thank you.
My cnvkit version:
Brad Chapman於 2018年2月2日星期五 UTC+8下午11時09分06秒寫道:
翰欽鄭
Feb 6, 2018, 6:57:40 AM2/6/18
to biovalidation
Hello:
I had examined my finished project.
And I have 2 questions listed below.
1. Lactobacillus pentosus is not listed in bcbio databases. Therefore, I use bcbio_setup_genome.py to add the reference into bcbio local database.
Because my annotation file format is NCBI Genbank, I use gb2genome.py (one python script in bcbio_nextgen) to transform it to gtf file.
However, I found that one of transformed gtf records seems to be wrong (figure below).
I guess that the program may not process the circular genome.
Therefore, please check is this caused by program bug or wrong operation.
The genbank format of CEW82_RS00005:
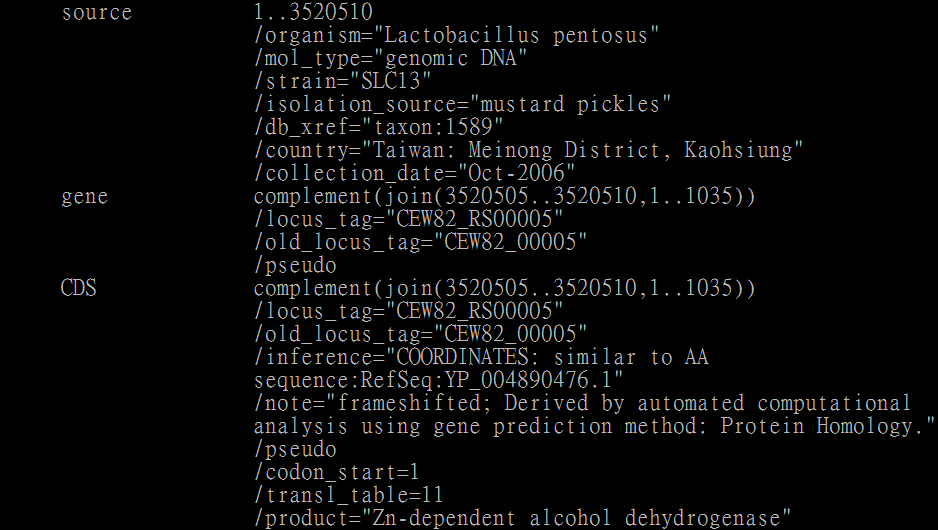
The gtf format of gene CEW82_RS00005

2. In variant calling output of bcbio, I expect that the vcf file can tell me variant with gene information.
But in the result, I didn't see it (figure below).
I want to ask whether gene information is provided in vcf file when I use custom genome.
Thank you.
The vcf output within in gene CEW82_RS00010:

CEW82_RS00010 region:

PS:
Genbank file link: links
The transformed gtf file link: links
I had examined my finished project.
And I have 2 questions listed below.
1. Lactobacillus pentosus is not listed in bcbio databases. Therefore, I use bcbio_setup_genome.py to add the reference into bcbio local database.
Because my annotation file format is NCBI Genbank, I use gb2genome.py (one python script in bcbio_nextgen) to transform it to gtf file.
However, I found that one of transformed gtf records seems to be wrong (figure below).
I guess that the program may not process the circular genome.
Therefore, please check is this caused by program bug or wrong operation.
The genbank format of CEW82_RS00005:
The gtf format of gene CEW82_RS00005
2. In variant calling output of bcbio, I expect that the vcf file can tell me variant with gene information.
But in the result, I didn't see it (figure below).
I want to ask whether gene information is provided in vcf file when I use custom genome.
Thank you.
The vcf output within in gene CEW82_RS00010:
CEW82_RS00010 region:
PS:
Genbank file link: links
The transformed gtf file link: links
The transformed fasta file link: links
The attach file is ensemble output of bcbio.
翰欽鄭於 2018年2月5日星期一 UTC+8下午1時45分12秒寫道:
翰欽鄭於 2018年2月5日星期一 UTC+8下午1時45分12秒寫道:
Brad Chapman
Feb 7, 2018, 9:11:27 AM2/7/18
to 翰欽鄭, biovalidation
Thanks for the feedback and suggestions.
You're right that the gb2genome.py file doesn't have any special provisions
for circular genomes. Apologies, you might need to do some post-processing to
correctly represent these. We'd be happy to contributions to help handle this
case better.
The gene information in the VCFs comes from snpEff (or VEP) annotations after
variant calling, so to use for your custom genome you'd have to download and
link into bcbio the relevant database. We don't have an automated process for
this now so this would have to be done manually and then referenced in the
configuration file. I've added some documentation about how to do this:
https://bcbio-nextgen.readthedocs.io/en/latest/contents/configuration.html#effects-prediction
Hope this helps with finalizing your analysis,
Brad
> [ text/plain ]
> Hello:
>
> I had examined my finished project.
> And I have 2 questions listed below.
>
> 1. *Lactobacillus pentosus* is not listed in bcbio databases. Therefore, I
>
> I had examined my finished project.
> And I have 2 questions listed below.
>
> use bcbio_setup_genome.py to add the reference into bcbio local database.
> Because my annotation file format is NCBI Genbank
> <https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP022130.1>, I use gb2genome.py
> Because my annotation file format is NCBI Genbank
> <https://github.com/chapmanb/bcbio-nextgen/blob/master/scripts/utils/gb2genome.py>
> (one python script in bcbio_nextgen) to transform it to gtf file.
> However, I found that one of transformed gtf records seems to be wrong
> (figure below).
> I guess that the program may not process the circular genome.
> Therefore, please check is this caused by program bug or wrong operation.
>
>
> The genbank format of CEW82_RS00005:
>
> However, I found that one of transformed gtf records seems to be wrong
> (figure below).
> I guess that the program may not process the circular genome.
> Therefore, please check is this caused by program bug or wrong operation.
>
>
> The genbank format of CEW82_RS00005:
>
> The gtf format of gene CEW82_RS00005
>
>
>
>
>
>
> 2. In variant calling output of bcbio, I expect that the vcf file can tell
> me variant with gene information.
> But in the result, I didn't see it (figure below).
> I want to ask whether gene information is provided in vcf file when I use
> custom genome.
>
> Thank you.
>
>
> The vcf output within in gene CEW82_RS00010:
>
> CEW82_RS00010 region:
> me variant with gene information.
> But in the result, I didn't see it (figure below).
> I want to ask whether gene information is provided in vcf file when I use
> custom genome.
>
> Thank you.
>
>
> The vcf output within in gene CEW82_RS00010:
>
>
>
> PS:
> Genbank file link: links
> <https://gist.github.com/blueskys123/7391900bbb72523f28e23002ff9af83f>
>
> PS:
> Genbank file link: links
> The transformed gtf file link: links
> <https://gist.github.com/blueskys123/769237ae58715bdf5429694d4bc2c42b>
> The transformed fasta file link: links
> <https://gist.github.com/blueskys123/123414da80f3dd5768666ddc69518e83>
> The attach file is ensemble output of bcbio.
>
> 翰欽鄭於 2018年2月5日星期一 UTC+8下午1時45分12秒寫道:
>>
>> Thank you for your comment.
>> I check my cnvkit version, and I found my cnvkit version is *py27_1*, not
>
> 翰欽鄭於 2018年2月5日星期一 UTC+8下午1時45分12秒寫道:
>>
>> Thank you for your comment.
>> py27_2.
>> Therefore, I updated my bcbio and reran the project.
>> The project works fine.
>>
>> Thank you.
>>
>> My cnvkit version:
>>
>>
>>
>>
>>
>> Therefore, I updated my bcbio and reran the project.
>> The project works fine.
>>
>> Thank you.
>>
>> My cnvkit version:
>>
>>
>>
>>
>>
> For more options, visit https://groups.google.com/d/optout.
> [ T-LA-F03_S17_PE_cutadapt_QC-ensemble-annotated-decomposed.vcf.gz: application/octet-stream ]
> [ Auto Generated Inline Image 1: image/png ]
> [ Auto Generated Inline Image 2: image/png ]
> [ Auto Generated Inline Image 3: image/png ]
> [ Auto Generated Inline Image 4: image/png ]
翰欽鄭
Feb 8, 2018, 4:19:29 AM2/8/18
to biovalidation
Thank you for your comment.
Because my strain, Lactobacillus pentosus SLC13, is not listed in snpEff database, I decide to build snpEff database and add it into Bcbio custom genome database.
Then, the analysis works fine.
I share my flow below. If you have any suggestion, please tell me.
Thank you.
1. download the genbank
2. check the genbank file. Make sure that the Sequence name in gb2genome.py translated fasta is same as CONTIG sequence name.
If the names are not same, the "ERROR_CHROMOSOME_NOT_FOUND" messages will display in vcf files.
(note: The gb2genome.py use NZ_CP022130.1 for sequence name)
Genbank file (Original header):
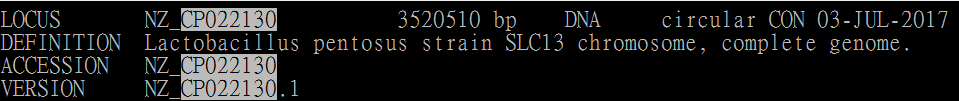
Genbank file (Original sequence header):

Genbank file (modified header, but I guess that there is not essential to modifiy here):

Genbank file (Modified sequence header):

3. Create snpEff database
Database name "Lactobacillus_pentosus_SLC13" is same as my build custom genome name.
Then modify the snpEff.config.
Open snpEff.config, add the words in figure, and save.
(If the species is not bacteria or plasmid, the "(database name).(Sequence name).codonTable : Baterial_and_Plant_Plastid" should not be required.)

Finally, build the snpeff database
If you want to check the chromosome name in build snpeff database, you can type the command:

4. add the snpeff database into bcbio
Then add the snpeff setting in seq/Lactobacillus_pentosus_SLC13-resources.yaml

Otherwise, the document has a typo:
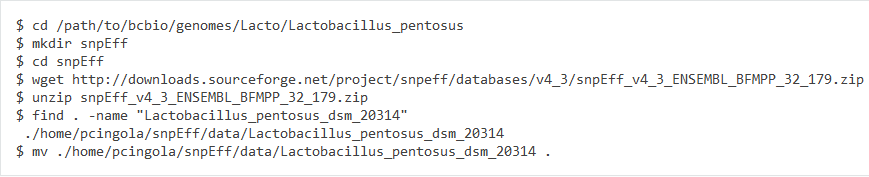
The create snpeff directory is "snpeff", not "snpEff".
Because I created snpEff and stored the snpeff data into this directory, I got an error.
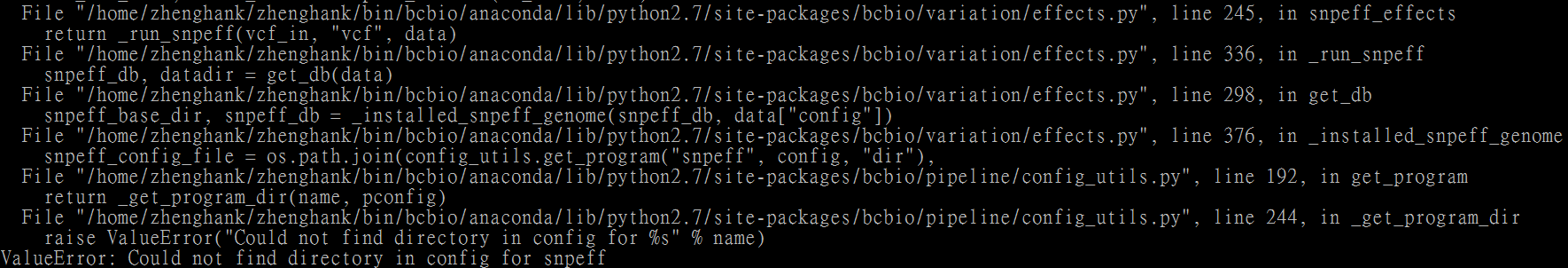
5. Run the analysis and you get the well annotated vcf file.

Brad Chapman於 2018年2月7日星期三 UTC+8下午10時11分27秒寫道:
Then, the analysis works fine.
I share my flow below. If you have any suggestion, please tell me.
Thank you.
1. download the genbank
2. check the genbank file. Make sure that the Sequence name in gb2genome.py translated fasta is same as CONTIG sequence name.
If the names are not same, the "ERROR_CHROMOSOME_NOT_FOUND" messages will display in vcf files.
(note: The gb2genome.py use NZ_CP022130.1 for sequence name)
Genbank file (Original header):
Genbank file (Original sequence header):
Genbank file (modified header, but I guess that there is not essential to modifiy here):
Genbank file (Modified sequence header):
3. Create snpEff database
cd /path/to/bcbio/anaconda/share/snpeff-4.3i-0
mkdir -p data/Lactobacillus_pentosus_SLC13
data/Lactobacillus_pentosus_SLC13
Database name "Lactobacillus_pentosus_SLC13" is same as my build custom genome name.
Then modify the snpEff.config.
Open snpEff.config, add the words in figure, and save.
(If the species is not bacteria or plasmid, the "(database name).(Sequence name).codonTable : Baterial_and_Plant_Plastid" should not be required.)
Finally, build the snpeff database
snpEff build -genbank -v Lactobacillus_pentosus_SLC13
If you want to check the chromosome name in build snpeff database, you can type the command:
snpEff Lactobacillus_pentosus_SLC13 -v4. add the snpeff database into bcbio
cd /path/to/bcbio/genomes/Lacto/Lactobacillus_pentosus_SLC13
mkdir snpeff
cd snpeff
mv /path/to/bcbio/anaconda/share/snpeff-4.3i-0/data/Lactobacillus_pentosus_SLC13 ./
Then add the snpeff setting in seq/Lactobacillus_pentosus_SLC13-resources.yaml
Otherwise, the document has a typo:
The create snpeff directory is "snpeff", not "snpEff".
Because I created snpEff and stored the snpeff data into this directory, I got an error.
5. Run the analysis and you get the well annotated vcf file.
Brad Chapman於 2018年2月7日星期三 UTC+8下午10時11分27秒寫道:
Brad Chapman
Feb 14, 2018, 7:35:50 AM2/14/18
to 翰欽鄭, biovalidation
Thanks much for following up and glad this worked for you. I added a pointer
to this discussion in the documentation as well so anyone facing similar
issues can learn from what you've done. Thanks again sharing this,
Brad
> Thank you for your comment.
> Because my strain, Lactobacillus pentosus SLC13, is not listed in snpEff
> database, I decide to build snpEff database and add it into Bcbio custom
> genome database.
> Then, the analysis works fine.
> I share my flow below. If you have any suggestion, please tell me.
> Thank you.
>
>
> 1. download the genbank
> 2. check the genbank file. Make sure that the Sequence name in gb2genome.py
> translated fasta is same as CONTIG sequence name.
> If the names are not same, the "ERROR_CHROMOSOME_NOT_FOUND" messages will
> display in vcf files.
> (note: The gb2genome.py use NZ_CP022130.1 for sequence name)
>
> Genbank file (Original header):
>
> Genbank file (Original sequence header):
>
>
>
>
> Genbank file (modified header, but I guess that there is not essential to
> modifiy here):
>
> modifiy here):
>
> Genbank file (Modified sequence header):
>
>
>
>
> 3. Create snpEff database
>
> cd /path/to/bcbio/anaconda/share/snpeff-4.3i-0
> mkdir -p data/Lactobacillus_pentosus_SLC13
> mv /path/of/modified/sequence.gb ./data/Lactobacillus_pentosus_SLC13/
> gene.gbk
>
> Database name "Lactobacillus_pentosus_SLC13" is same as my build custom
> genome name.
> Then modify the snpEff.config.
>
> Open snpEff.config, add the words in figure, and save.
> (If the species is not bacteria or plasmid, the "(database name).(Sequence
> name).codonTable : Baterial_and_Plant_Plastid" should not be required.)
>
>
>
> cd /path/to/bcbio/anaconda/share/snpeff-4.3i-0
> mkdir -p data/Lactobacillus_pentosus_SLC13
> mv /path/of/modified/sequence.gb ./data/Lactobacillus_pentosus_SLC13/
> gene.gbk
>
> Database name "Lactobacillus_pentosus_SLC13" is same as my build custom
> genome name.
> Then modify the snpEff.config.
>
> Open snpEff.config, add the words in figure, and save.
> (If the species is not bacteria or plasmid, the "(database name).(Sequence
> name).codonTable : Baterial_and_Plant_Plastid" should not be required.)
>
>
> Finally, build the snpeff database
> snpEff build -genbank -v Lactobacillus_pentosus_SLC13
>
>
> If you want to check the chromosome name in build snpeff database, you can
> type the command:
> snpEff Lactobacillus_pentosus_SLC13 -v
>
>
>
> snpEff build -genbank -v Lactobacillus_pentosus_SLC13
>
>
> If you want to check the chromosome name in build snpeff database, you can
> type the command:
> snpEff Lactobacillus_pentosus_SLC13 -v
>
>
>
> 4. add the snpeff database into bcbio
>
> cd /path/to/bcbio/genomes/Lacto/Lactobacillus_pentosus_SLC13
> mkdir snpeff
> cd snpeff
> mv /path/to/bcbio/anaconda/share/snpeff-4.3i-0/data/Lactobacillus_pentosus_SLC13 ./
>
>
>
>
> Then add the snpeff setting in
> seq/Lactobacillus_pentosus_SLC13-resources.yaml
>
>
>
> cd /path/to/bcbio/genomes/Lacto/Lactobacillus_pentosus_SLC13
> mkdir snpeff
> cd snpeff
> mv /path/to/bcbio/anaconda/share/snpeff-4.3i-0/data/Lactobacillus_pentosus_SLC13 ./
>
>
>
>
> Then add the snpeff setting in
> seq/Lactobacillus_pentosus_SLC13-resources.yaml
>
>
> Otherwise, the document has a typo:
>
> The create snpeff directory is "*snpeff*", not "*snpEff*".
>
> Because I created snpEff and stored the snpeff data into this directory, I
> got an error.
>
>
>
> got an error.
>
>
>
> 5. Run the analysis and you get the well annotated vcf file.
>
>
>
>
>
>
>> > To post to this group, send email to bioval...@googlegroups.com
>> <javascript:>.
>> > Visit this group at https://groups.google.com/group/biovalidation.
>> > To view this discussion on the web visit
>> https://groups.google.com/d/msgid/biovalidation/cc331c1e-f331-4d3d-9466-32b44edf831a%40googlegroups.com.
>>
>> > For more options, visit https://groups.google.com/d/optout.
>> > [ T-LA-F03_S17_PE_cutadapt_QC-ensemble-annotated-decomposed.vcf.gz:
>> application/octet-stream ]
>> > [ Auto Generated Inline Image 1: image/png ]
>> > [ Auto Generated Inline Image 2: image/png ]
>> > [ Auto Generated Inline Image 3: image/png ]
>> > [ Auto Generated Inline Image 4: image/png ]
>>
>
>> > To view this discussion on the web visit
>> https://groups.google.com/d/msgid/biovalidation/cc331c1e-f331-4d3d-9466-32b44edf831a%40googlegroups.com.
>>
>> > For more options, visit https://groups.google.com/d/optout.
>> > [ T-LA-F03_S17_PE_cutadapt_QC-ensemble-annotated-decomposed.vcf.gz:
>> application/octet-stream ]
>> > [ Auto Generated Inline Image 1: image/png ]
>> > [ Auto Generated Inline Image 2: image/png ]
>> > [ Auto Generated Inline Image 3: image/png ]
>> > [ Auto Generated Inline Image 4: image/png ]
>>
>
> --
> You received this message because you are subscribed to the Google Groups "biovalidation" group.
> To unsubscribe from this group and stop receiving emails from it, send an email to biovalidatio...@googlegroups.com.
> To post to this group, send email to bioval...@googlegroups.com.
> Visit this group at https://groups.google.com/group/biovalidation.
> To view this discussion on the web visit https://groups.google.com/d/msgid/biovalidation/652054bb-d048-458a-98fd-07b4243b0ba6%40googlegroups.com.
> You received this message because you are subscribed to the Google Groups "biovalidation" group.
> To unsubscribe from this group and stop receiving emails from it, send an email to biovalidatio...@googlegroups.com.
> To post to this group, send email to bioval...@googlegroups.com.
> Visit this group at https://groups.google.com/group/biovalidation.
Reply all
Reply to author
Forward
0 new messages