recent blog posts on randomisations in Biodiverse

Shawn Laffan
observed patterns against some expectation based on randomly shuffling
the observed data.
Biodiverse supports a range of randomisation algorithms, and Version 4
will support the Independent Swaps algorithm. In Version 4 it will also
be possible to calculate z-scores of the distributions in addition to
the rank-relative scores.
To provide more details about the new features and randomisations in
general, I have uploaded a series of blog posts over November and December.
Changes to how Cluster and RegionGrower analyses are randomised in v4:
https://biodiverse-analysis-software.blogspot.com/2020/11/updated-handling-of-cluster-and-region.html
Z-score calculations:
https://biodiverse-analysis-software.blogspot.com/2020/11/randomisations-now-also-generate-z.html
How the rand_structured randomisation works:
https://biodiverse-analysis-software.blogspot.com/2020/11/randomisations-how-randstructured.html
Modelling spatial structure in the randomisations:
https://biodiverse-analysis-software.blogspot.com/2020/11/randomisations-modelling-spatial.html
Spatially partitioning randomisations into subsets:
https://biodiverse-analysis-software.blogspot.com/2020/11/spatially-partition-your-randomisations.html
The independent swaps algorithm, including an alternate implementation
and a comparison with the rand_structured algorithm:
https://biodiverse-analysis-software.blogspot.com/2020/12/biodiverse-now-includes-independent.html
If you want to read other blog posts about the randomisations in
Biodiverse then they are grouped under a tag and can be accessed through
this URL:
https://biodiverse-analysis-software.blogspot.com/search/label/randomisations
Comments on the posts are disabled due to spam, but feel free to ask
questions on this mailing list.
Regards,
Shawn.

Gabriela Procópio Camacho
(https://biodiverse-analysis-software.blogspot.com/2020/11/spatially-partition-your-randomisations.html) but I've been wondering how would I define the subsets exactly? For example, I have a shape file with the ecoregions definitions for my area of study, and would like to run randomisations given those ecoregions, but how could I enter this information on Biodiverse?
Shawn Laffan
The help system needs a lot of work so any questions are always welcome.
The condition to use for this is sp_points_in_same_poly_shape
https://github.com/shawnlaffan/biodiverse/wiki/SpatialConditions#sp_points_in_same_poly_shape
The help system needs an example but you can use it like this (adjusting the path as needed):
sp_points_in_same_poly_shape (file => 'C:\path\to\shapefile')
What this will do is create a set of basedata objects, with one basedata object for each ecoregion. The labels and groups in each ecoregion will then be randomised separately, and then reassembled into one basedata object for the actual calculations and comparisons.
The main thing to watch for is that the test for if a group is in an ecoregion is based on the centroid of the group. This means it is possible for a group to be allocated to a very small ecoregion if that ecoregion contains the group's centroid. I usually work around this issue by generalising the shapefile in some way before using it.
If you have groups near the edges of the ecoregion polygons then you might also encounter issue 780 if you are using version 3.1 or earlier. This is fixed in the current development release (version 3.99_001).
https://github.com/shawnlaffan/biodiverse/issues/780
Regards,
Shawn.
--
You received this message because you are subscribed to the Google Groups "Biodiverse Users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to biodiverse-use...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/ba766032-a9f7-4175-9ed4-fead9021df9cn%40googlegroups.com.
-- Prof Shawn Laffan, FMSSANZ School of Biological, Earth and Environmental Sciences UNSW, Sydney 2052, Australia Tel +61 2 9065 5607 https://www.bees.unsw.edu.au/our-people/shawn-laffan https://shawnlaffan.github.io/biodiverse (free diversity analysis software) International Journal of Geographical Information Science http://www.tandf.co.uk/journals/ijgis UNSW CRICOS Provider Code 00098G
Gabriela Procópio Camacho
Gabriela P. Camacho, Ph.D.
Postdoctoral Fellow
Pronouns: she/her/hers
Center for Integrative Biodiversity Discovery
Museum für Naturkunde
T +49 30 889140 8592
gabriela.camacho@mfn.berlin
Leibniz-Institute for Evolution and Biodiversity Science
Invalidenstr. 43
10115 Berlin
Germany
www.museumfuernaturkunde.berlin

To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/e2a63f93-d014-86f1-7263-ace4f03a4f11%40unsw.edu.au.
Shawn Laffan
That shapefile could be used, but it needs to be filtered to only contain the polygons you want to use.
Identify the names of the branches you want from the tree and extract those polygons from the current shapefile to a new shapefile. This can be done using a table selection.
Branches can be identified by hovering on the tree in Biodiverse and looking for the node label above the map. You can also control-click on them to get more of the details under the NODE_VALUES list.
The shapefile contains repeated entries for each field of interest so it would also be useful to also select only features where the KEY field is equal to TOTAL_LENGTH (any of them will do, really).
The reason this repetition exists is that shapefiles have a limit of 11 characters for field names. Future versions of Biodiverse will support other formats that do not have this limit.
If you are not sure how to do it then I can provide some guidance. Just let me know which system you are using (e.g. ArcGIS, QGIS, R spatial tools, ...).
And as an aside, this whole selection process will be avoidable when the code for issue 757 is implemented. It is currently on the list for version 4.
https://github.com/shawnlaffan/biodiverse/issues/757
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/CA%2B%3DicGKJLPoh_6kTw9gScGuX8HzNQSUiTmemA66R0PLT-M5_xA%40mail.gmail.com.
Gabriela Procópio Camacho
Gabriela P. Camacho, Ph.D.
Postdoctoral Fellow
Pronouns: she/her/hers
Center for Integrative Biodiversity Discovery
Museum für Naturkunde
T +49 30 889140 8592
gabriela.camacho@mfn.berlin
Leibniz-Institute for Evolution and Biodiversity Science
Invalidenstr. 43
10115 Berlin
Germany
www.museumfuernaturkunde.berlin
You received this message because you are subscribed to a topic in the Google Groups "Biodiverse Users" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/biodiverse-users/DIb0NomF6rU/unsubscribe.
To unsubscribe from this group and all its topics, send an email to biodiverse-use...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/4e2ba2fb-c9ed-d6e1-6d6a-784db4bb7b74%40unsw.edu.au.
Gabriela Procópio Camacho
Gabriela P. Camacho, Ph.D.
Postdoctoral Fellow
Pronouns: she/her/hers
Center for Integrative Biodiversity Discovery
Museum für Naturkunde
T +49 30 889140 8592
gabriela.camacho@mfn.berlin
Leibniz-Institute for Evolution and Biodiversity Science
Invalidenstr. 43
10115 Berlin
Germany
www.museumfuernaturkunde.berlin
Shawn Laffan
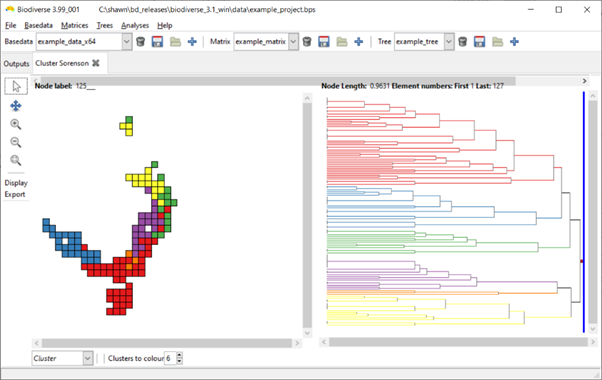
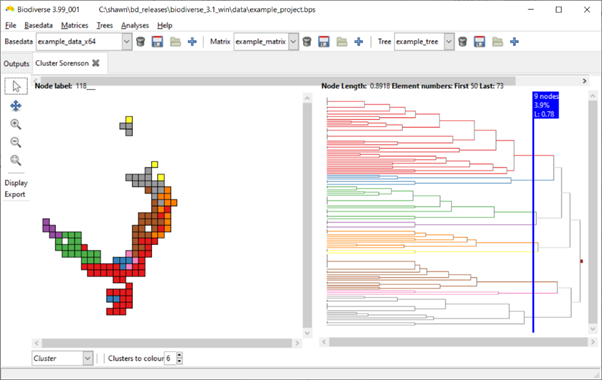
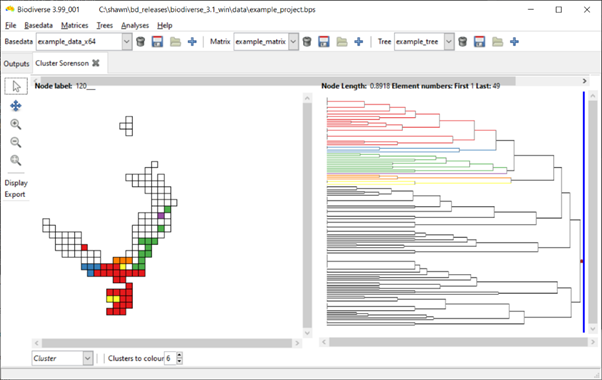
The default in the output is to show six clusters. This is an arbitrary number and there is no analysis behind it.
It is hard to define an optimal cutoff. One of the benefits of the trees is that they show nested groups. Examples are at https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0092558 and also Fig 3 of https://onlinelibrary.wiley.com/doi/10.1111/ddi.12129
I would suggest you try using the slider to see what effect different cutoffs have on the patterns. That will stop colouring unique values after 13 clusters as there are not enough colours in the palette, but you can also click on individual branches to see within them.
Some example screenshots are below (hopefully not too low resolution). The first is the default display with six clusters. In the second the slider has been moved across nine branches (nodes), so nine clusters are coloured. In the third screenshot, one of the branches has been clicked on and six sub-clusters are coloured.
The "clusters to colour" option at the bottom of the window determines how many clusters are coloured when branches are clicked on. The slider does not use this value.
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/CA%2B%3DicGKW06u7cSe%3Dvj3sb9tgJFkb_s8ovD0nFuC4875NYRWUcg%40mail.gmail.com.
Shawn Laffan
A tutorial for selections in QGIS is at https://www.qgistutorials.com/en/docs/3/working_with_attributes.html
I'll see what I can work up for the Biodiverse specific aspects, although it might be easier for in the long term me to implement the code for issue 757 and avoid the selection gymnastics in the first place.
With that in mind I've started work on issue 757 so the next development version will make this process easier. I'm hoping to release that soon.
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/CA%2B%3DicGJOcCsFFFoP1%2BZQwRcPVAD8aMxO4u_KQq5KGbV3SbwuDg%40mail.gmail.com.
Shawn Laffan
Given your use case is to use clusters, I decided to add some spatial conditions that allow a user to directly check if groups are in a subcluster (sp_point_in_cluster and sp_points_in_same_cluster).
These are in the 3.99_002 development release. Could you give them a try and let me know if they help?
In terms of what to use, if you want to define spatial regions in an analysis, or subregions for a spatially constrained randomisation, then use sp_points_in_same_cluster. If you want to use a subcluster as a definition query then use sp_point_in_cluster.
Some examples are below. In each of them, "some_cluster_output" is the name of the cluster analysis you want to use.
Regards,
Shawn.
## sp_points_in_same_cluster examples
# Try to use the highest four clusters from the root.
# Note that the next highest number will be used
# if four is not possible, e.g. there might be five
# siblings below the root. Fewer will be returned
# if the tree has insufficient tips.
sp_points_in_same_cluster (
output => "some_cluster_output",
num_clusters => 4,
)
# Cut the tree at a distance of 0.25 from the tips
sp_points_in_same_cluster (
output => "some_cluster_output",
target_distance => 0.25,
)
# Cut the tree at a depth of 3 from the root.
# The root is depth 1.
sp_points_in_same_cluster (
output => "some_cluster_output",
target_distance => 3,
group_by_depth => 1,
)
# work from an arbitrary node
sp_points_in_same_cluster (
output => "some_cluster_output",
num_clusters => 4,
from_node => '118___', # use the node's name
)
# target_distance is ignored if num_clusters is set
sp_points_in_same_cluster (
output => "some_cluster_output",
num_clusters => 4,
target_distance => 0.25,
)
## sp_point_in_cluster examples
# This will select any element that is a terminal in the cluster output
# It is useful if the cluster analysis was run under
# a definition query and you want the same set of elements.
sp_point_in_cluster (
output => "some_cluster_output",
)
# Now specify a cluster within the output
sp_point_in_cluster (
output => "some_cluster_output",
from_node => '118___', # use the node's name
)
# Specify an element to check instead of the current
# processing element.
sp_point_in_cluster (
output => "some_cluster_output",
from_node => '118___', # use the node's name
element => '123:456', # specify an element to check
)
Gabriela Procópio Camacho
Gabriela P. Camacho, Ph.D.
Postdoctoral Fellow
Pronouns: she/her/hers
Center for Integrative Biodiversity Discovery
Museum für Naturkunde
T +49 30 889140 8592
gabriela.camacho@mfn.berlin
Leibniz-Institute for Evolution and Biodiversity Science
Invalidenstr. 43
10115 Berlin
Germany
www.museumfuernaturkunde.berlin
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/31f85f9c-67a8-c860-2310-611917664f47%40unsw.edu.au.
Shawn Laffan
Can you please send me the exact error message? And the condition you are using?
Also, are you using version 3.1 or one of the development versions like 3.99_002? There is a related issue that is fixed in the dev versions.
https://github.com/shawnlaffan/biodiverse/issues/780
If it is simpler, and you are willing, then you can also send me the bidiverse project file. This can be direct to my email so it does not go to the list. If the file is larger than 5MB then we can use a file sender (I can provide an upload voucher using http://cloudtstor.aarnet.edu.au if needed). If you do send the file then I will not use it for any purpose other than debugging this issue.
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/CA%2B%3DicG%2BB_CuVuJO-iKW0PKCgjZUJGcu8676HJL9VBJPd8%2BWp9g%40mail.gmail.com.