Correct usage of codon models with rate heterogeneity across sites and mX(a) models estimating omega.

Karolis Ramanauskas
I have been running and comparing the output of various codon models with and without rate heterogeneity. Additionally, I have been attempting to test for positive selection by passing the omega parameter to mX(a)_test models. Below is a selection of model combinations I have running, alphabet="Codons", imodel="rs07" in all cases; smodel definitions are as follows:
1. gtr+fMutSel
2. gtr+fMutSel+Rates.gamma[4]
3. function[w,gtr+fMutSel[w]]+m8a_test
4. function[w,gtr+fMutSel[w]]+m8a_test+Rates.gamma[4]
I have also tried gy94 and mg94 (and their variants) instead of fMutSel. I have noticed an apparently significant difference in the estimates of the Rates.gamma:alpha parameter between the models 2 and 4 (currently 1.768 and 6.734, respectively) which does not look right. I understand that the ordering of functions determines how they are interpreted and nested by BAli-Phy. Did I specify the models incorrectly? My intention was to apply the discrete gamma distribution of rates between codon sites, but I am concerned that this may not be the case in model 4.

Benjamin Redelings
Hi Karolis,
Ah, nice to see you are experimenting with interesting models!
Your models 2 and 4 below are not equivalent, so the Rates.gamma:alpha values cannot really be expected to have the same value. To explain in more detail:
2. gtr+fMutSel+Rates.gamma[4]This a 4-component mixture, where different components run at different rates.
In this model, Rates.gamma:alpha describes differences in conservation AND differences in the synonymous rate ....... which are assumed to be the same thing.
This is not a completely realistic model because it will use rate
variation to explain more conserved codons. That means that, for
more conserved codons, the 3rd codon position will also evolve
slower, which should not be the case.
3. function[w,gtr+fMutSel[w]]+m8a_test
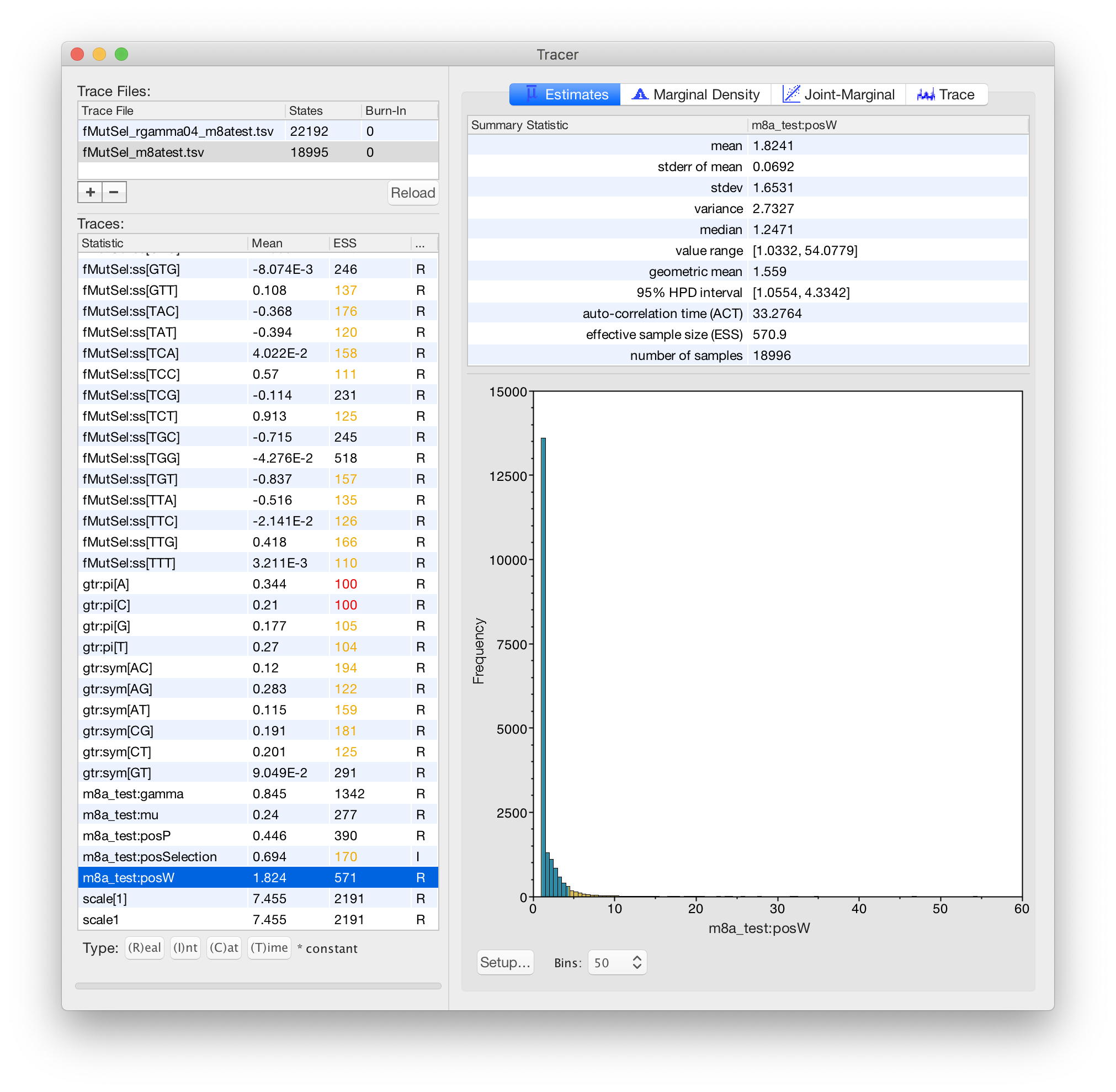
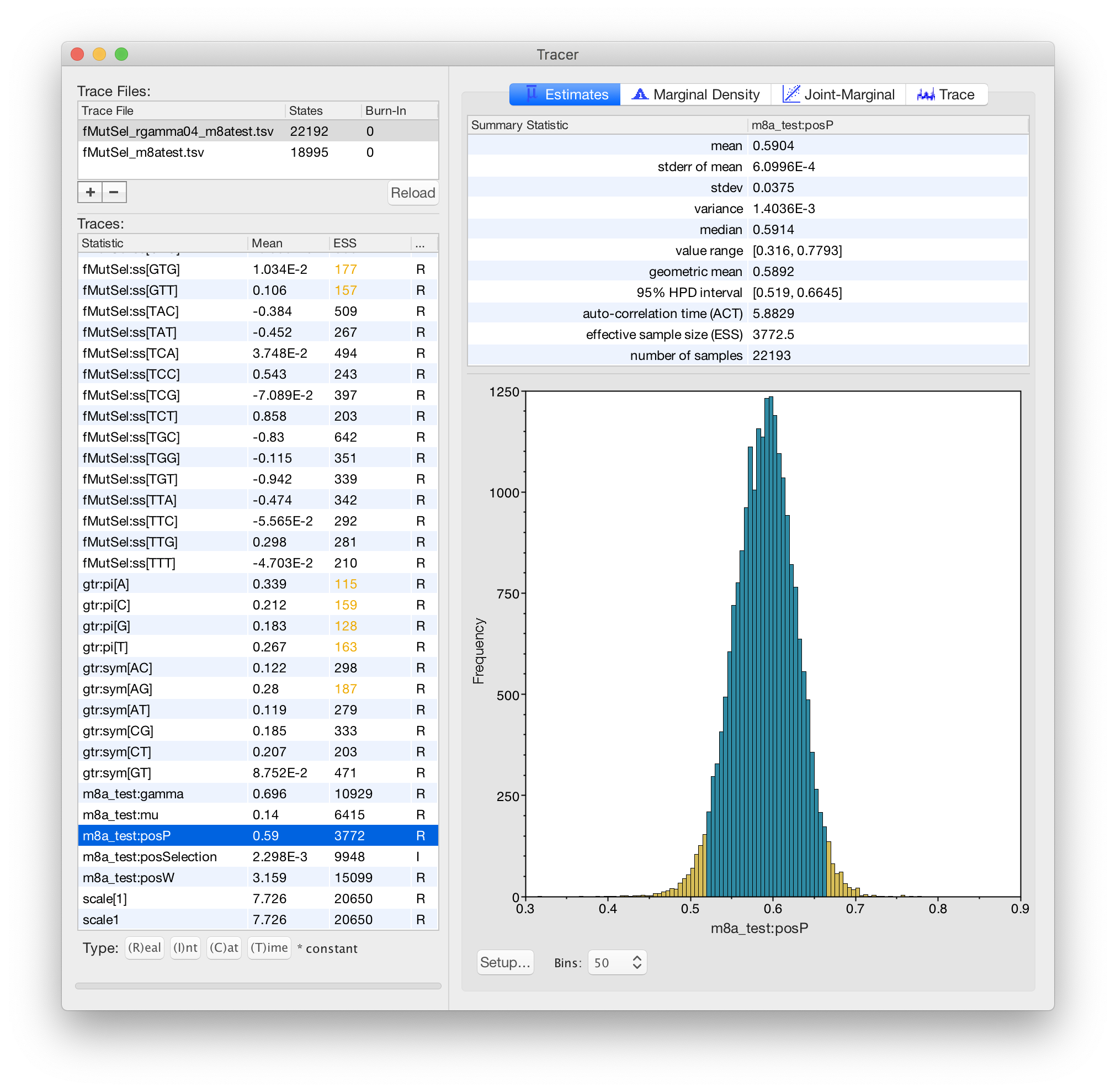
This is also a 4-component mixture, but different components have different omega values instead of different rates.
In this model, m8a_test:gamma describes differences in
*conservation*.
This is more realistic than your model #2, because the more-conserved components will change amino-acids at a slower rate, but the 3rd codon position will not evolve slower at more conserved sites.
4. function[w,gtr+fMutSel[w]]+m8a_test+Rates.gamma[4]
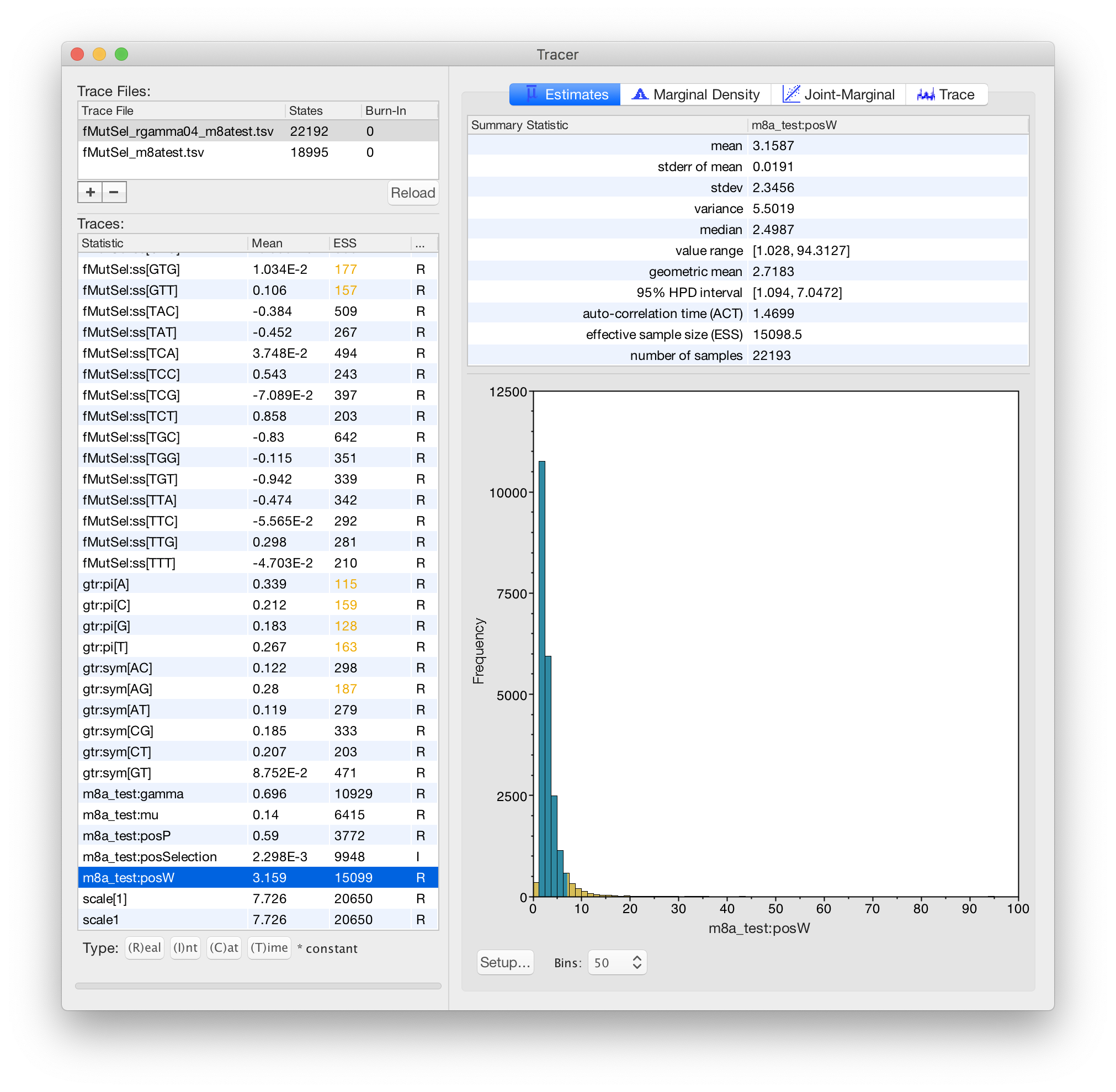
This is a 16-component mixture. Mixture components have BOTH different dN/dS values AND different rates.
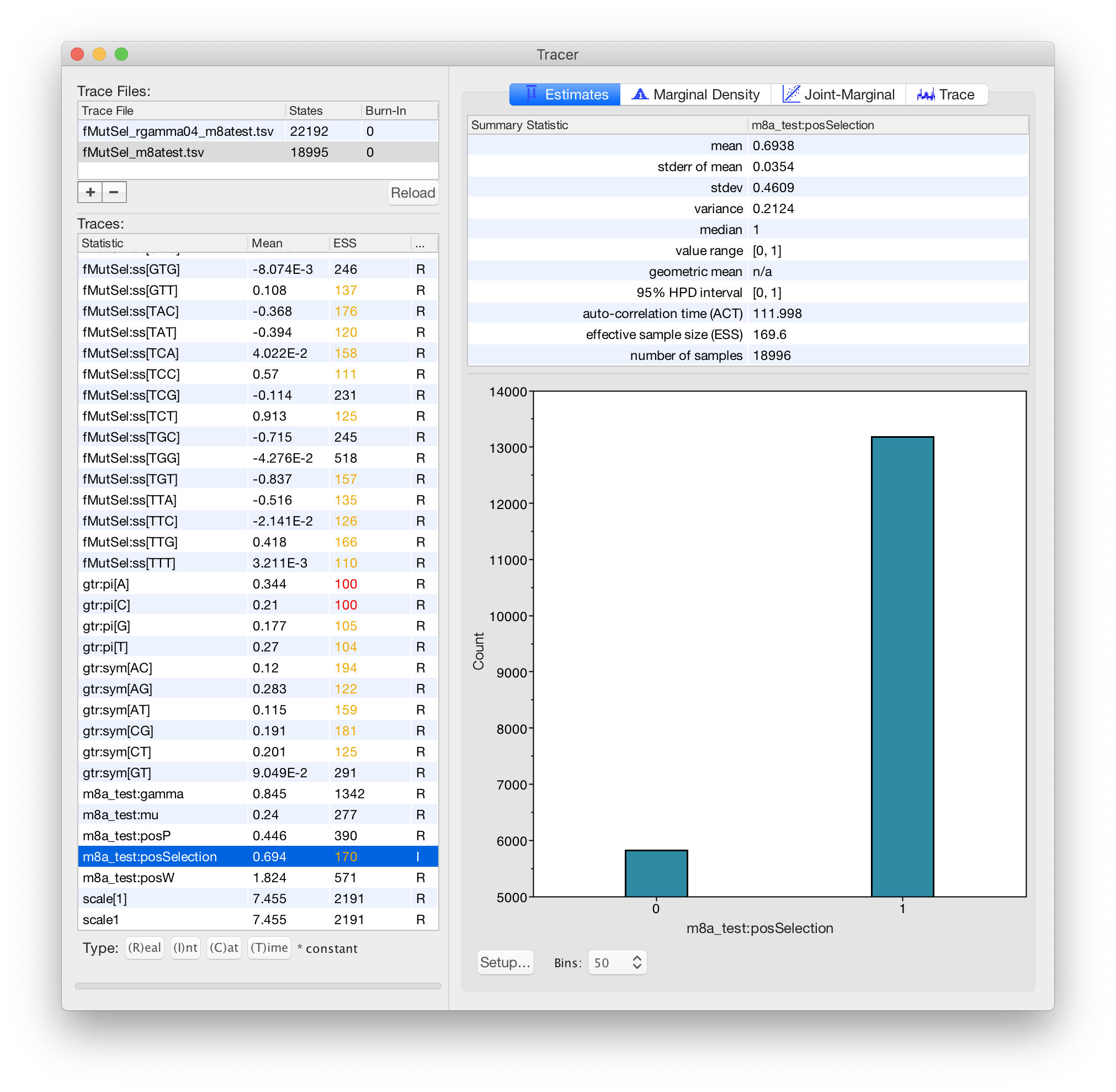
In this model, m8a_test:gamma describes differences in *conservation*, and Rates.gamma:alpha describes differences in the *synonymous rate*.
This model has both different omega values AND different rates. This basically extends model 3 to allow differences in the SYNONYMOUS rate at different codons, in addition to different ratios between the synonymous and non-synonymous rates.
In this model, the Rates.gamma:alpha is NOT responsible for describing differences in conservation. The m8a_test part of the model is responsible for describing differences in conservation. So the Rates.gamma:alpha value is expected to be higher (indicating less variation) than in your #2 model, because it is not handling variation in conservation, but only variation in the synonymous rate.
Does that make sense?
-BenRI
P.S. You subscribed to the list with your gmail address, but since you have been sending messages from your UIC address, this might explain why you are not getting replies. (Also messages are not getting sent until I approve them). Maybe you should subscribe both addresses, but disable e-mail delivery for one of them? Not completely sure...
P.P.S. When I tested out these models, I used fMutSel0, which
can be a bit faster. It has fewer parameters to estimate, since
it does not allow different codons for the same amino acid to have
different fitnesses.
--
You received this message because you are subscribed to the Google Groups "bali-phy-users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to bali-phy-user...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/bali-phy-users/5dad3436-2eb5-4649-a575-66fcb650bb2cn%40googlegroups.com.
Karolis Ramanauskas

Benjamin Redelings
To view this discussion on the web visit https://groups.google.com/d/msgid/bali-phy-users/c6cf91f6-45c0-4f31-b528-7967656a7098n%40googlegroups.com.