CBS input dataframe
59 views
Skip to first unread message

Emanuel Gonçalves
Jun 20, 2017, 1:05:48 PM6/20/17
to aroma.affymetrix
Dear all,
I'm processing a set of ~200 SNP6 .cel files and then performing segmentation with CBS. But I would like to know how to extract the data points data-frame that is used as input to DNAcopy CBS.
The code is as below:
# install.packages('aroma.affymetrix', repos='http://cran.us.r-project.org')
library("aroma.affymetrix")
# Logs
log <- verbose <- Arguments$getVerbose(-8, timestamp=TRUE)
# Reduce decimal places to minimize space
options(digits=4)
print('Options configured')
# -- SNP6 cancer cell lines preprocessing with CRMA (v2)
gsT <- doCRMAv2("cancer_lines", chipType="GenomeWideSNP_6,Full", verbose=verbose)
print('cancer_lines CRMAv2 done.')
# -- Segmentation (CBS)
sm <- CbsModel(gsT)
fit(sm, verbose=verbose)
print('CbsModel done.')
# -- Export segmentation
pathname <- writeRegions(sm, verbose=verbose)
print('Exported done.')
I would like to extract the gsT as a data.frame in the format that is used in DNAcopy:
> library(DNAcopy)
> data(coriell)
> head(coriell)
Clone Chromosome Position Coriell.05296 Coriell.13330
1 GS1-232B23 1 0 NA 0.207470
2 RP11-82d16 1 468 0.008824 0.063076
3 RP11-62m23 1 2241 -0.000890 0.123881
4 RP11-60j11 1 4504 0.075875 0.154343
5 RP11-111O05 1 5440 0.017303 -0.043890
6 RP11-51b04 1 7000 -0.006770 0.094144
Thank you in advance,

Henrik Bengtsson
Jun 20, 2017, 4:51:35 PM6/20/17
to aroma-affymetrix
Hi.
There's no direct way of doing this, but as a start you can read in
all the writeRegions()-exported segment data for all samples in a data
set as explained in
http://www.aroma-project.org/vignettes/NonPairedCBS/. That will give
you a data.frame.
From there you should be able to manipulate the content of the
data.frame using basic R tools, e.g. subset(), rbind(), cbind() etc.
The dplyr package might be a more convenient way of doing it - it's
the ninja-tool for data.frame manipulations.
Hope this help
Henrik
PS. Feel free to share your solution here.
> --
> --
> When reporting problems on aroma.affymetrix, make sure 1) to run the latest
> version of the package, 2) to report the output of sessionInfo() and
> traceback(), and 3) to post a complete code example.
>
>
> You received this message because you are subscribed to the Google Groups
> "aroma.affymetrix" group with website http://www.aroma-project.org/.
> To post to this group, send email to aroma-af...@googlegroups.com
> To unsubscribe and other options, go to http://www.aroma-project.org/forum/
>
> ---
> You received this message because you are subscribed to the Google Groups
> "aroma.affymetrix" group.
> To unsubscribe from this group and stop receiving emails from it, send an
> email to aroma-affymetr...@googlegroups.com.
> For more options, visit https://groups.google.com/d/optout.
There's no direct way of doing this, but as a start you can read in
all the writeRegions()-exported segment data for all samples in a data
set as explained in
http://www.aroma-project.org/vignettes/NonPairedCBS/. That will give
you a data.frame.
From there you should be able to manipulate the content of the
data.frame using basic R tools, e.g. subset(), rbind(), cbind() etc.
The dplyr package might be a more convenient way of doing it - it's
the ninja-tool for data.frame manipulations.
Hope this help
Henrik
PS. Feel free to share your solution here.
> --
> When reporting problems on aroma.affymetrix, make sure 1) to run the latest
> version of the package, 2) to report the output of sessionInfo() and
> traceback(), and 3) to post a complete code example.
>
>
> You received this message because you are subscribed to the Google Groups
> "aroma.affymetrix" group with website http://www.aroma-project.org/.
> To post to this group, send email to aroma-af...@googlegroups.com
> To unsubscribe and other options, go to http://www.aroma-project.org/forum/
>
> ---
> You received this message because you are subscribed to the Google Groups
> "aroma.affymetrix" group.
> To unsubscribe from this group and stop receiving emails from it, send an
> email to aroma-affymetr...@googlegroups.com.
> For more options, visit https://groups.google.com/d/optout.
Emanuel Gonçalves
Jun 20, 2017, 5:33:36 PM6/20/17
to aroma.affymetrix
Hi Henrik,
There's no direct way of doing this, but as a start you can read in
all the writeRegions()-exported segment data for all samples in a data
set as explained in
http://www.aroma-project.org/vignettes/NonPairedCBS/. That will give
you a data.frame.
But that would get me the segmentation regions, right? What I want is to export the input of CBS.
> # -- SNP6 cancer cell lines preprocessing with CRMA (v2)
> gsT <- doCRMAv2("cancer_lines", chipType="GenomeWideSNP_6,Full",
> verbose=verbose)
> print('cancer_lines CRMAv2 done.')
In other words, is there a writeRegions function for "gsT" to export it as a table with the probe, chromosome, position and the ratios per sample.
Thank you for your time,
Emanuel Gonçalves
Jun 20, 2017, 7:40:07 PM6/20/17
to aroma.affymetrix
I think what I'm looking for is:
extractRawCopyNumbers(cbsmodel, array=1, chromosome=2)
Problem is I have ~200 and the function seems to be taking quite sometime to get me the results (is this because I don't have all the cbs computed yet?), is there a way to export the whole data-set raw copy numbers?
Thank you,
Henrik Bengtsson
Jun 21, 2017, 12:15:20 AM6/21/17
to aroma-affymetrix
Ah, my bad - you're correct.
On Tue, Jun 20, 2017 at 4:40 PM, Emanuel Gonçalves
<emanuelv...@gmail.com> wrote:
> I think what I'm looking for is:
>
>> extractRawCopyNumbers(cbsmodel, array=1, chromosome=2)
>
>
> Problem is I have ~200 and the function seems to be taking quite sometime to
> get me the results (is this because I don't have all the cbs computed yet?),
> is there a way to export the whole data-set raw copy numbers?
Yes, that's the way to do it.
That's what fit() for CbsModel is using internally, and yes, there's a
bit of overhead in each of those calls. There's quite a bit of
validation etc going on. It's probably possible to make it faster for
the case when one would want to pull out data for all samples and all
chromsomes at once - but, as far as I remember, I don't think we
implemented that per se. If you want to do your own segmentation, see
example at the end.
These day you can parallelize it quite easily using future, cf.
http://www.aroma-project.org/howtos/parallel_processing/. For
example, here's how you can extract chromosomes in parallel:
future::plan("multiprocess")
array <- 1L
cns <- future_lapply(1:23, FUN = function(chr) {
extractRawCopyNumbers(cbsmodel, array = array, chromosome = chr)
})
If you have completed fit(), then all the CBS results (segments and
locus-level data) are saved to file for each (array, chromosome).
These "internal" data files are located in:
path <- getPath(sm)
You can pull out, say, all Chr 1 results across all samples as:
pathnames <- dir(path = getPath(sm), pattern = ",chr01,", full.names = TRUE)
fits <- lapply(pathnames, FUN = loadObject)
locusData <- lapply(fits, FUN = `[[`, "data")
However, those files don't contain information of the cell names
("clones" aka "probe names").
Below is a way you can get the normalized locus-level CN signals that
can be used for downstream segmentation. This example uses
Mapping10K_Xba142 data, but the idea is the same:
> clones <- getUnitNames(getUnitNamesFile(gsT))
> positions <- readDataFrame(getAromaUgpFile(gsT))
> signals <- extractMatrix(gsT)
> data <- cbind(clones, positions, signals)
> str(data)
'data.frame': 10208 obs. of 13 variables:
$ clones : Factor w/ 10208 levels "AFFX-5Q-123",..: 1 2 3 4 5013
9020 3048 8172 4263 10008 ...
$ chromosome: int NA NA NA NA 6 7 10 1 15 12 ...
$ position : int NA NA NA NA 162491313 42608794 68113302 22754354
28848178 53641300 ...
$ GSM226867 : num 5209 2042 5005 3750 1563 ...
$ GSM226868 : num 4799 1995 4726 3719 1675 ...
$ GSM226869 : num 5069 2070 4661 3450 1881 ...
$ GSM226870 : num 5502 2492 5164 3809 1828 ...
$ GSM226871 : num 5462 2298 4917 3678 2131 ...
$ GSM226872 : num 5232 1886 4671 3459 2227 ...
$ GSM226873 : num 5878 2479 5588 4372 1817 ...
$ GSM226874 : num 6363 2668 5768 4356 1396 ...
$ GSM226875 : num 4789 1765 4543 3610 1876 ...
$ GSM226876 : num 4762 1876 4614 3241 2568 ...
With those data, you can run your own segmentation - but you do have
to worry about how calculate CN ratios, i.e. what is T and what is N
in C = T / N etc.
Hope this helps
Henrik
On Tue, Jun 20, 2017 at 4:40 PM, Emanuel Gonçalves
<emanuelv...@gmail.com> wrote:
> I think what I'm looking for is:
>
>> extractRawCopyNumbers(cbsmodel, array=1, chromosome=2)
>
>
> Problem is I have ~200 and the function seems to be taking quite sometime to
> get me the results (is this because I don't have all the cbs computed yet?),
> is there a way to export the whole data-set raw copy numbers?
That's what fit() for CbsModel is using internally, and yes, there's a
bit of overhead in each of those calls. There's quite a bit of
validation etc going on. It's probably possible to make it faster for
the case when one would want to pull out data for all samples and all
chromsomes at once - but, as far as I remember, I don't think we
implemented that per se. If you want to do your own segmentation, see
example at the end.
These day you can parallelize it quite easily using future, cf.
http://www.aroma-project.org/howtos/parallel_processing/. For
example, here's how you can extract chromosomes in parallel:
future::plan("multiprocess")
array <- 1L
cns <- future_lapply(1:23, FUN = function(chr) {
extractRawCopyNumbers(cbsmodel, array = array, chromosome = chr)
})
If you have completed fit(), then all the CBS results (segments and
locus-level data) are saved to file for each (array, chromosome).
These "internal" data files are located in:
path <- getPath(sm)
You can pull out, say, all Chr 1 results across all samples as:
pathnames <- dir(path = getPath(sm), pattern = ",chr01,", full.names = TRUE)
fits <- lapply(pathnames, FUN = loadObject)
locusData <- lapply(fits, FUN = `[[`, "data")
However, those files don't contain information of the cell names
("clones" aka "probe names").
Below is a way you can get the normalized locus-level CN signals that
can be used for downstream segmentation. This example uses
Mapping10K_Xba142 data, but the idea is the same:
> clones <- getUnitNames(getUnitNamesFile(gsT))
> positions <- readDataFrame(getAromaUgpFile(gsT))
> signals <- extractMatrix(gsT)
> data <- cbind(clones, positions, signals)
> str(data)
'data.frame': 10208 obs. of 13 variables:
$ clones : Factor w/ 10208 levels "AFFX-5Q-123",..: 1 2 3 4 5013
9020 3048 8172 4263 10008 ...
$ chromosome: int NA NA NA NA 6 7 10 1 15 12 ...
$ position : int NA NA NA NA 162491313 42608794 68113302 22754354
28848178 53641300 ...
$ GSM226867 : num 5209 2042 5005 3750 1563 ...
$ GSM226868 : num 4799 1995 4726 3719 1675 ...
$ GSM226869 : num 5069 2070 4661 3450 1881 ...
$ GSM226870 : num 5502 2492 5164 3809 1828 ...
$ GSM226871 : num 5462 2298 4917 3678 2131 ...
$ GSM226872 : num 5232 1886 4671 3459 2227 ...
$ GSM226873 : num 5878 2479 5588 4372 1817 ...
$ GSM226874 : num 6363 2668 5768 4356 1396 ...
$ GSM226875 : num 4789 1765 4543 3610 1876 ...
$ GSM226876 : num 4762 1876 4614 3241 2568 ...
With those data, you can run your own segmentation - but you do have
to worry about how calculate CN ratios, i.e. what is T and what is N
in C = T / N etc.
Hope this helps
Henrik
Emanuel Gonçalves
Jun 21, 2017, 5:56:56 AM6/21/17
to aroma.affymetrix
Hi Henrik,
Perfect with your help I putted together a script that seems capable of extracting the raw counts (over a pooled reference):
# - Importslibrary("Biobase")library("aroma.affymetrix")
# - Reduce decimal places to minimize space
options(digits=4)
# - Load datagsT <- doCRMAv2("cancer_lines", chipType="GenomeWideSNP_6,Full", verbose=verbose)print('cancer_lines CRMAv2 done.')
# - Get the genome information fileclones <- getUnitNames(getUnitNamesFile(gsT))
positions <- readDataFrame(getAromaUgpFile(gsT))row.names(positions) <- clones
# - Calculate raw counts with pooled referencetheta <- extractMatrix(gsT)rownames(theta) <- clones
thetaR <- rowMedians(theta, na.rm=TRUE)
M <- log2(theta / thetaR)row.names(M) <- clones
# - Exportpositions <- positions[complete.cases(positions), ]M <- M[rownames(positions), ]
write.table(positions, './raw_total_cn_pooled_positions.txt', sep='\t', quote=F)write.table(M, './raw_total_cn_pooled.txt', sep='\t', quote=F)
Actually, I have seen this effect also using as reference the average of a batch (~250 samples) of SNP6 normal samples. My end goal is to obtain GISTIC scores and when I fed the CbsModel output to GISTIC I noticed that there are more '-1's than '0's, again showing there is a skew to negative ratios.
Am I missing any preprocessing that is required besides the doCRMAv2?
Thank you for your help,
Thank you for your help,
{kind=link}
Emanuel Gonçalves
Jun 22, 2017, 5:35:19 PM6/22/17
to aroma.affymetrix
Hi,
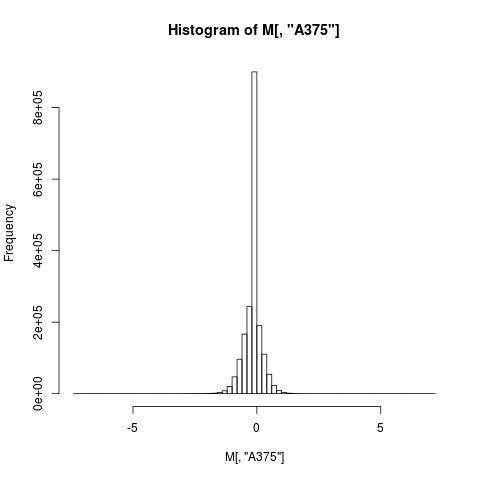
This seems to be working well, although I'm concerned with the raw counts distributions of some cell lines as they look skewed to negative values (histogram attached, apologises for the poor quality).Actually, I have seen this effect also using as reference the average of a batch (~250 samples) of SNP6 normal samples. My end goal is to obtain GISTIC scores and when I fed the CbsModel output to GISTIC I noticed that there are more '-1's than '0's, again showing there is a skew to negative ratios.Am I missing any preprocessing that is required besides the doCRMAv2?
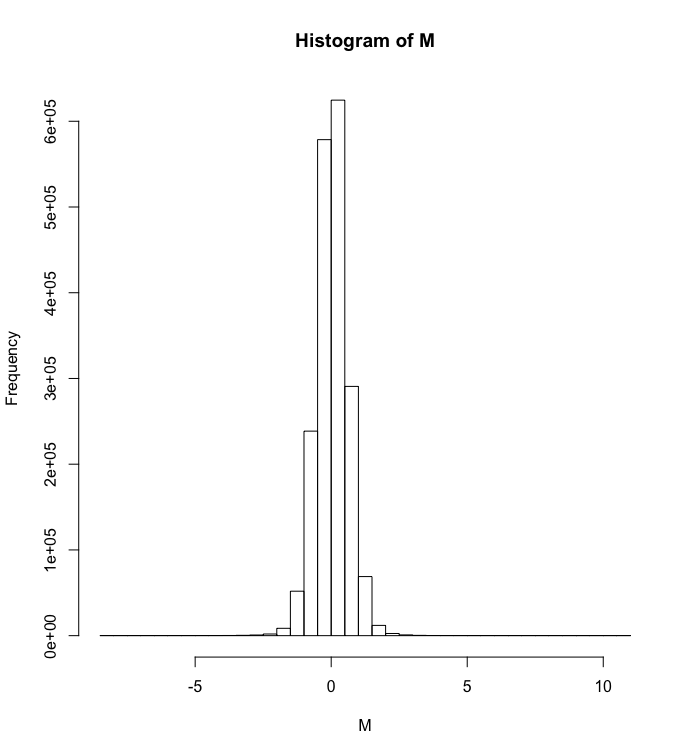
Just to follow up, if I follow the vignette CRMAv2 with all the steps (http://aroma-project.org/vignettes/CRMAv2/) the histogram of the ratios is now centred on zero, attached. Is doing step-by-step slower than 'doCRMAv2'? It seemed to be taking more time, but I haven't calculated the differences.
In the vignette code:
if (length(findUnitsTodo(plm1)) > 0) { # Fit CN probes quickly (~5-10s/array + some overhead) units1 <- fitCnProbes(plm1, verbose=verbose) str(units1) # int [1:945826] 935590 935591 935592 935593 935594 935595 ... # Fit remaining units, i.e. SNPs (~5-10min/array) units1 <- fit(plm1, verbose=verbose) str(units1)}Is not processing the SNPs an option if you are only interested in copy-number changes? That would be ideal :)
Thanks,
{kind=link}
Reply all
Reply to author
Forward
0 new messages